"Goal 1 -- Identify and Characterize the Molecular Machines of Life – the Multiprotein (Multimolecule) Complexes that Execute Cellular Functions and Govern Cell Form"
The importance of peptide tandem MS data relative to other methods:
If one thinks broadly about the methods available for "complexes", a dozen potential methods (and various combinations) leap to the front as foundations for augmenting homology/literature based models. These include: (a) two-hybrid assays (Steffen et al. 2002*), (b) in vitro assays (Bulyk et al 2001, 2002), (c) antibody/protein arrays (Mcbeath et al. 2001), (d) in vivo fluorescent tag microscopy, (e) fluorescence resonance energy transfer (FRET) (Ting et al. 2001), (f) whole protein-MS (Smith et al. 2002), (g) 2Dgels (Link et al. 1997; Grunenfelder et al. 2001) , (h) in vitro crosslinking, (i) electron microscopy/ crystallography/ NMR, (j) native complex fractionation (Link et al. 1997), (k) peptide-MS (Chen et al 2001; Link; Li et al 2001), (l) in vivo crosslinking (Hartemink et al. 2002; Wyrick et al. 2001; Steffen et al 2002b* see attached). We have experience with nearly all of these methods but will focus on the last three (j-l) in this grant for the following reasons. Some (e,i) are not convincingly poised to be high-throughput enough to monitor a variety of environment effects. The problem with many light microscopy-based methods (d) is that the spatial resolution is too coarse for the smallest microbial cells. Two-hybrid data and in vitro pairwise interactions miss interactions that require larger numbers of interacting proteins and nucleic acids. Furthermore, whole protein-protein interaction data (f) is not as useful as peptide-peptide interactions for docking structures and/or determining possible competing interactions. Peptide data are also preferable to even the most precise protein masses for establishing the phosphorylation state of each peptide site. (Stemmann 2001, Ficarro et al. 2002).
Goal 1a: Proteogenomic Mapping
Proper analysis of mass spectrometric data from peptide fragments can lead to enormous insights into the primary structure of an organism's genome. We have developed "proteogenomic mapping" as a technique to represent information about proteins and peptides discovered through mass spectrometry on a genome-based scaffold. Using this technique, we were able to re-assign proper boundaries to ORFs and discover new ORFs in many instances in Mycoplasma pneumoniae (Jaffe, et al 2002, *see attached). We were also able to identify certain ORFs that are likely to be bogus in the current annotation. Application of these methods to the target organisms will more accurately define their genome structures and our understanding of how they operate.
This is possible through multidimensional chromatography of proteins and peptides coupled to tandem mass spectrometry. The current plateau using 2-dimensional chromatography of peptides (cation exchange and reverse phase) is about 1500 proteins per experiment (Washburn et al. 2001). Using in-house modifications of this technique we were able to cover > 80% of expected open reading frames (ORFs) of a small bacterium (Mycoplasma pneumoniae) with a genome on the order of 1 Mb. However, additional dimensions of chromatographic separations may be added upstream at the protein level to provide both resolution and information. We will incorporate upstream separations by Size-Exclusion Chromatography (SEC) and Ion EXchange chromatography (IEX) on native proteins prior to proteolytic digestion and further separation of the peptides. In addition to complexity reduction, these separation techniques contribute information about the size and isoelectric properties of the analytes. (see Goal 1b)
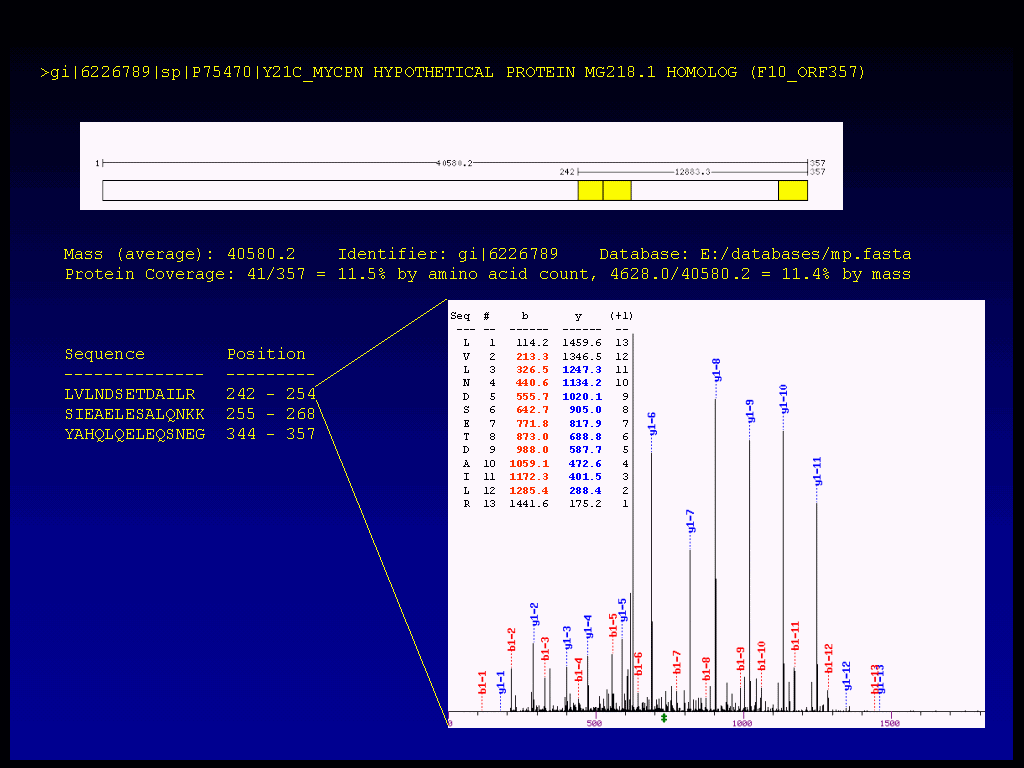
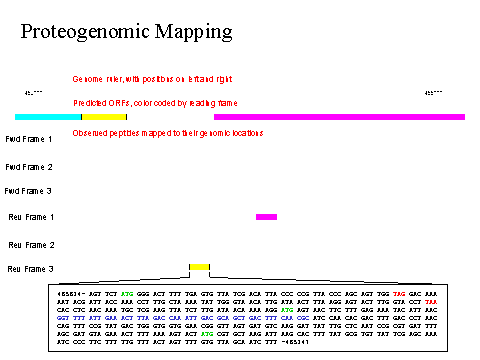
Figure 1a. Illustration of multidimensional MS data and of the software for proteogenomic mapping. Note the importance of using all six possible reading frames rather than relying on the genome annotation. Note that the raw MS data behind all assertions are hypertext linked.
The above figure 1a is a close-up view of all six frames of 1 kbp of the M. pneumoniae proteogenome showing the hypertext connections to the relevant DNA sequences and primary MS2 mass data.
Figure 1b (below), in contrast, shows a full-genome view for an initial sampling of Prochlorococcus peptides. The usual option for stop codon display has been suppressed for these illustrations.
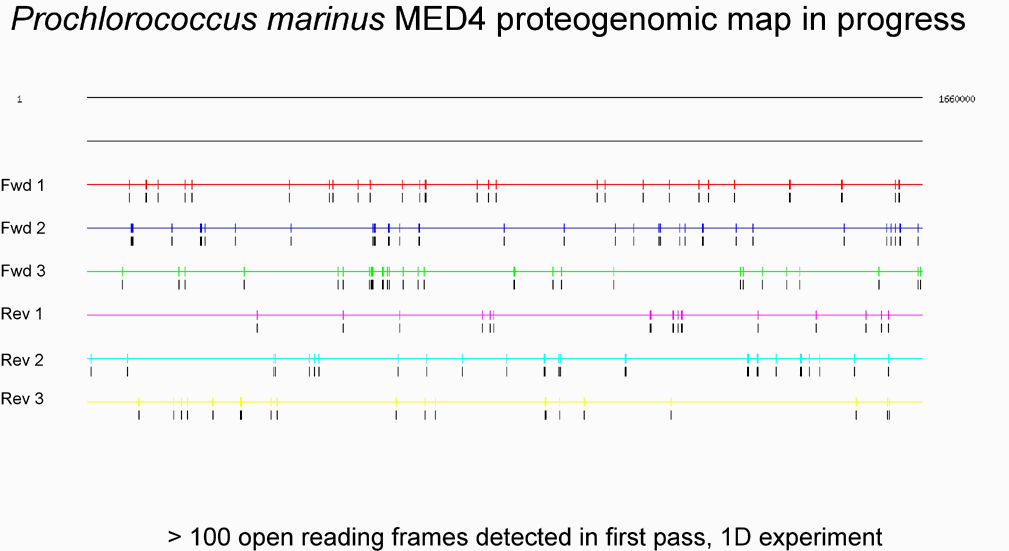
We have already developed protocols and software that have allowed us to measure over 80% of the predicted proteins (without use of tags) in a bacterial cell population (Mycoplasma pneumoniae) including subcellular localization and post-synthetic modifications (phosphorylation & proteolysis). We would propose to move this closer to 100% and larger proteomes. We estimate that we can analyze up to 1500 proteins using 4-dimensions for peptides (Cation exchange LC, Reverse-phase LC, MS, MS2) and at least 10-fold more using additional protein size exclusion LC (SEC).
Our community now has 5 operational Finnigan ion-trap mass-spectrometers and 75 Linux CPU nodes. These are fully loaded and generally not available to microbial proteomics of the type described in the proposal. Based on our experience with the Mycoplasma proteome pipeline we expect that we will need 10, 20 and 40 Linux CPU nodes per Ion-trap MS instrument for Prochlorococcus, Caulobacter, and Pseudomonas, respectively, for analysis with SEQUEST-PVM (Eng et al., 1994; Tabb, et al, 2001; http://www.csm.ornl.gov/pvm/). It should be noted that this version of SEQUEST is more scalable than the commercial version and that we are one of only 4 labs outside of the Yates lab that have access to it due to restrictions by Finnigan. We will need 6 additional nodes (relatively independent of proteome size) for our multidimensional separation quantitation software (Leptos & Church 2002 in prep. see attached*). This is a total of 180 Linux nodes on average to match the capacity of the mass spectrometers. Based on the above, with 5 ion-traps available, we expect to be able to complete data collection in 1 week for each Prochlorococcus light/dark/cell-cycle proteome native subcellular fraction, (2 weeks for Caulobacter), and 3 weeks for Pseudomonas. For time-series data we anticipate that we may be able to focuse the MS time on the MS spectra and quantitation displacing some of the MS2 and its associated costs will be shifted to the quantitation and modeling tasks, for which scalling costs are still in flux.
One of our first priorities will be to determine the time-resolution necessary for data collection in terms of statistical significance between adjacent time points and biological significance as indicated by modeling simulations. For example, in a bacterial transcriptional regulatory network, the rate of transcription and translation can occur at about 50 nucleotides per second, and full-length proteins can be synthesized, folded and functional within 30 seconds. Therefore, current rates of sampling in biological experiments will probably not be sufficient for our computer modeling goals of investigating cause and effect relationships. For example, the 15 minute sampling interval used by Laub et al. 2000*, is theoretically sufficient for 30 distinct regulatory steps to occur, even mediated by the relatively slow process of transcriptional control. Even a sampling rate consistent with rates of transcription may be too coarse relative to the rapid regulatory steps that can occur in postsynthetic protein modifications, e.g. proteolysis and phosphorylation. On the other hand, there is no need to collect data more finely than the resolution that the technical specifics of obtaining synchronized cell populations permits. This will be addressed by comparing three of the best systems for cell division synchronization: Caulobacter by natural solid phase to motile asymmetric division, Prochlorococcus by circadian entrainment, and Saccharomyces by rapid temperature-shift of a conditional cdc mutant (the Saccharomyces part of the project will be done mainly form other funding sources, however the data will be useful for comparing different synchronization efficiencies to fully survey current practical limits). We will begin by taking an interval suspected of having rapid proteomic changes from previous (coarse) time-series and successively divide the interval in half until no statistically significant differences can be observed between adjacent time points. We will also examine time-courses not involving cell-cycle that might display more synchrony, e.g. heat-shock. For our studies on time-series of antibiotic effects on bacterial RNA profiles (Cheung et al 2002*), we developed simple methods for rapidly collecting time points from pressurized chemostat growth vessels directly into lysis buffers or low temperatures. An interval as short as 2 seconds between samples can be achieved. One of the computational challenges is to optimally align duplicate and quasi-duplicate time-series that may have non-linear warping of the time axes due to natural and experimental processes. We have preliminary results on an algorithm to accomplish this (Aach and Church 2001*). We expect that the performance will be greatly improved with more smoothly varying curves produced by more finely sampled time courses.
Another goal is more comprehensive detection and interpretation of the mass spec peptides. While this is not common practice, we feel that it will be crucial to biosystems modeling efforts which are often sensitive to missing components . We will take each of the major mass peaks (in decreasing order of abundance) that are unexplained by current software and attempt to resolve the source of the peak. This type of analysis has the potential to identify new amino-acid modifications (biological and/or chemical) and resolve collision-induced dissociation (CID) prior to the expected MS2 CID step in combination with peptide charge. We will explore the properties of the predicted peptides as to their observed abundance in the spectrum in an attempt to get higher percent detection than our current average of 30%. The ionization (and hence detection) seems to be weakest for peptides with more acidic groups. MeOH/HCl esterification of the Asp, Glu and C-termini, appears to be a significant improvement in the missing-peptides problem and probably improves sensitivity overall since every peptide has at least one carboxyl group. Detection is also weak for phosphopeptides (probably for similar reasons), enrichment for these using Immobilized Metal Affinity Chromatography IMAC (Porath & Olin 1983; Conlon & Murphy 1976) has recently become much more feasible in part because of improvements above which also impact the IMAC adsorption (Ficarro, et al. 2002). Another option would be EDC coupling of acidic resiudes to cationic amines.
Goal 1b: Quantitative Interaction Proteomics
We will perform "Quantitative Proteomics" to extend the list of translated gene products from goal 1a to their relative and/or absolute abundances in the cell under various conditions in the target organisms. This is possible through extension of the multidimensional chromatography of peptides above in goal 1a to proteins. Additional dimensions of chromatographic separations may be added upstream to provide both resolution and information. We will incorporate upstream separations by Size-Exclusion Chromatography (SEC) and Ion EXchange chromatography (IEX) on native proteins prior to proteolytic digestion and further separation of the peptides. In addition to complexity reduction, these separation techniques contribute information about the size and isoelectric properties of the analytes, which in turn facilitates modeling of multi-protein complex composition.
Quantitation of protein products will be performed directly by algorithms currently under development in our laboratories. (Leptos & Church 2002 in prep. see attached*). We will also explore stable isotope incorporation techniques analogous to Isotope-Coded Affinity Tags (ICAT) (Gygi 1999) and Absolute QUAntitation (AQUA) (Stemmann 2001, Gygi et al unpublished). We will utilize our resource of purified heterologously expressed proteins to provide a nearly full-proteome set of standards for quantitation calibration and method development. The development of cloning, sequence confirmation and highly-parallel tag-purification methods is a separately funded ongoing collaboration between our group (Nick Reppas) and the Harvard Institute for Proteomics (Director Josh LaBaer). In analogy to the studies that we have done and will do on RNA stability (Selinger et al. 2002, see attached*) we will measure protein stability in the presence of protein synthesis inhibitors. We will also use stable isotope pulse-chase as a check. These decay parameters will be helpful in modeling done in Goal 4.
Goal 1c: Proteomic Cellular Interaction Deconvolution
We will use various biochemical techniques to isolate proteins from their local cellular environments to generate a high-resolution map of the target organisms. For instance, we have developed methods akin to chromatin immunoprecipitation (Hartemink et al 2002;Wyricket al. 2001; Laub MT et al. 2002*) to determine DNA-binding proteins en masse so that we may accurately place certain proteins physically on the chromosome (Steffen, et al. 2002b* see attached). These methods take advantage of the fact that formaldehyde will crosslink 12 of the 20 main amino acid side-chains as well as the peptide backbone for all 20 (French & Edsall,1945). We are also developing methods to determine the set of expressed membrane proteins using solid-phase cell-surface derivatization techniques, and secreted proteins should be easily obtained by simple biochemistry. Moreover, we can either passively scan or specifically target cellular proteins for alternative post-translational modification states such as phosphorylation and methylation. Simple peptide derivatization techniques coupled with immobilized metal affinity chromatography (IMAC) have been shown to generate a strong enrichment for phosphopeptides from a complex mixture (Ficarro et al 2002). Finally, we will trap native protein-protein interactions and enrich for them through the use of affinity-tagged cross-linking reagents such as Sulfo-SBED (Pierce, Inc.). Taken together, these powerful biochemical techniques coupled with mass spectrometry will allow us to draw a detailed map of the locations, states, and interactions of the proteins which compose cellular systems. It is also important to note the importance of being peptide-oriented as in Aims 1 and 2 since it helps to determine which segments of proteins interact. This is a clear advantage over methods such a 2D gels that measure only the mass of proteins.
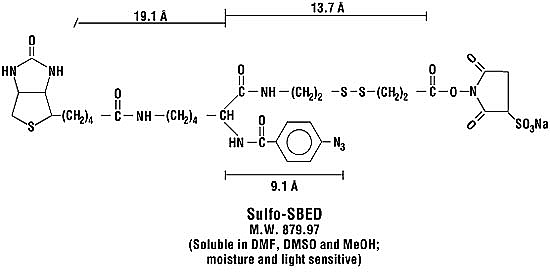
Figure 1c. The four reactive groups are (1) Sulfo-NHS ester, a amine group-specific reactivity (typically lysine) , (2) phenyl azide, nonspecific photoreactivity, (3) the biotin handle allows enrichment for peptides which have reacted (via avidin/streptavidin/NeutrAvidin solid phase methods), and (4) thiol cleavable. Feature 4 allows "reduction" of mass spectra derived with two N-termini (Chen et al 2001*)
http://www.piercenet.com/Products/Browse.cfm?fldID=02030410#
For crosslinking, a significant problem is half-links. Using Strong Cation Exchange columns (SCE) one can collect the >= +4 peak due to two alpha-aminos plus two Lys/Arg termini. Only single peptides with more than one His and/or LysPro and/or ArgPro will be co-elute, but these can occur in higher abundance than the crosslinked peptides. A condensing agent (such as 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide, EDC) avoids half-links, but is not general as it works on salt-bridges (Asp/Glu with Lys) and has only the single SCE selection. The Pierce triple-agent (see Figure 1c, above) looks like a promising solution in combination with SCE. Since the peptides would have to pass both the SCE >= +4 and the biotin selections efficiently.