"Goal 2 -- Characterize Gene Regulatory Networks"
Our initial focus will be on MED4, the high-light adapted strain of Prochlorococcus, because it has the smallest genome of the two sequenced ecotypes. We will analyze its responses to a set of well-defined experimental perturbations (light, temperature, and carbon) to help us begin to understand and construct a model of their cellular architectures. We also propose to determine how such responses change in the presence of members of the natural community. Transcriptional, translational and post-translational responses will be analyzed, to identify as completely as possible, the stimulons associated with each perturbation. Selected analyses will also be carried out on MIT9313, the low-light adapted ecotype, following our conviction of the power of a comparative approach. One of our foci is to understand the regulatory functions that direct the response of the cell to environmental change ¾ i.e. the cascade of events that bring about acclimation of metabolism to new conditions. This includes identifying the molecules and proteins involved in sensing and transmitting messages, the modulons and regulons associated with these regulators, the interactions of regulators with promoter sites, and the mechanism of the phenotypic change associated with these molecular responses.
BACKGROUND AND PROGRESS TO DATE - Goal 2
Environment/Gene Interactions: Key Environmental Variables for Prochlorococcus
As a phototroph, Prochlorococcus thrives in the oceans using a minimal and well-defined set of resources. Thus it is not difficult to design experiments that will test the dominant environmental perturbations that a cell might encounter. Analyses of a cell’s complete transcriptional and translational program in response to such perturbations will provide new perspectives on its regulatory networks by revealing genes that are co-regulated, and post-translational modifications that modify protein activity across a variety of conditions. Analyses across a variety of environmental variables, and comparison of the two ecotypes, will provide insights on the inter-relationship between pathways and processes. It is also likely to help in identifying regulatory proteins that link metabolic pathways and may expose presently unknown links. In this proposal we will be focusing on light, temperature, and carbon acquisition, and the interaction with heterotrophs and bacteriophage, thus we review here what is known about the genes involved in dealing with these variables in Prochlorococcus.
Light: Photoadaptation and Photoacclimation
One important difference between Prochlorococcus and other cyanobacteria is the presence of only one copy of psbD, which encodes the Photosystem II reaction center polypeptide D2. Furthermore, MED4 also possesses only one copy of psbA (which codes for D1) while MIT9313 has two copies, which encode identical polypeptides. Multiple gene copy numbers and iso-forms of these proteins directly affect the ability of the reaction centers to respond to light stress and photoinhibition . As shown above the two ecotypes of Prochlorococcus have distinct differences in light optima and in their chl b2/chl a2 ratios (Fig. 1 A,B). They also differ in the number of genes encoding the major light- harvesting chl-binding protein (pcb). MED4 possesses only one pcb gene, while MIT9313 has two and the low light-adapted isolate SS120 has as many as seven . In studies of acclimated steady-state cultures of MED4 psbA transcript levels were always higher at high irradiances . In cells grown on light/dark cycles, photosystem and antenna protein genes exhibit very different rhythms . In particular, the pcbA mRNA, shows two peaks and two minima (i.e. two complete cycles within 24 h). This is very different from the well-studied circadian expression patterns of light-harvesting proteins of virtually all other organisms, both prokaryotes and eukaryotes . Therefore it is relevant to elucidate whether and the extent to which these coordinated changes differ between the two ecotypes and what their functional implications are.
Despite its overall reduced genome size, MED4 has as many as 21 genes encoding putative high-light-inducible proteins (HLIP's) while MIT9313 has only nine putative HLIP genes. Although it has been suggested that HLIP's may be involved in photoprotection, their exact role is still unknown. In Synechococcus PCC7942, the expression of a closely related gene, hliA, is strongly induced by high irradiances or UV/blue light . In Synechocystis PCC6803, the levels of all five Hli polypeptides were found to be elevated in high light, and three of these proteins were also elevated in response to other stresses. (He, 2001 #1041). These results clearly point to the necessity of a combination of methods both at the RNA and protein level, including gene tagging and gene knock-outs if possible to address the role of Hli proteins.
In contrast to other cyanobacteria (including Synechocystis), neither of the Prochlorococcus genomes contain known photoreceptor genes, such as those encoding phytochromes, which have very important functions in cyanobacteria: CikA in Synechococcus elongatus serves to reset the clock in response to light , RcaE in Fremyella diplosiphon is critical for complementary chromatic adaptation , and Synechocystis Cph1 appears to be a taxis receptor . Thus how might Prochlorococcus sense light? One candidate for a light sensor in Prochlorococcus is phycoerythrin, which has been evolutionarily retained in the genome of MED4, as a highly derived single b phycoerythrin gene .
The use of microarrays to examine the expression of the many HLIP-like genes in MED4 and MIT9313 will enable us to establish whether the transcription of these genes is enhanced under specific light conditions, and to understand further their possible role in photoacclimation. The application of proteomics analysis (Goal 1) will show whether fluctuations in mRNA steady state levels are translated into corresponding changes in protein abundance, and a more in-depth analysis of the phycoerythrin function might unravel a novel function for this well-known pigment.
Cell Synchrony induced by light-dark cycles: One of the key advantages of the Prochlorococcus system for studying cellular networks is that the cell division cycle synchronizes beautifully when grown on a light/dark cycle . This has been well documented in the laboratory as well as the field (Figure 2a, below). It is unknown if cell division is regulated by a circadian clock, but it is intriguing that MED4 and MIT9313 contain homologues to two of the three components of the clock in Synechococcus PCC7942 (kaiB and kaiC).
Carbon Metabolism
Inorganic Carbon: Prochlorococcus MED4 and MIT9313 possess one contiguous stretch of genes involved in carbon assimilation that was likely obtained by horizontal gene transfer (HGT) from purple bacteria. The gene order ¾ csoS1A(ccmK)-rbcLS-csoS2-csoS3-orfA-orfB in MED4 and csoS1A(ccmK)-rbcLS-csoS2-csoS3-orfA-orfB-csoS1A(ccmK) in MIT9313 ¾ is highly similar to that found in chemoautotrophs such as Thiobacillus. Some of these genes have more (csoS1A to ccmK) or less (orfA and orfB to ccmL) homology to genes known to be involved in the cyanobacterial carbon concentrating mechanism (CCM). The important role of Rubisco in carboxysome assembly makes it plausible that the entire CCM and Rubisco complex in Prochlorococcus (and marine Synechococcus) was acquired by HGT. Whether this variant CCM provides an ecologically significant advantage in acquiring CO2 remains to be seen.
An efficient CCM requires the active uptake of inorganic carbon in the form of CO2 and/or HCO3- and the creation of an elevated local CO2 concentration within the carboxysome, in close proximity to Rubisco . In cyanobacteria, carbonic anhydrase, which is associated with carboxysomes , generates CO2 from the accumulated HCO3-. Carbonic anhydrase exists in three distinct classes, and is widespread in metabolically diverse species from both the Archaea and Bacteria. Its role is particularly well investigated in cyanobacterial CO2 fixation , thus it is all the more surprising that neither of the Prochlorococcus genomes contains a gene with homology to any of the known carbonic anhydrases. Moreover, there are no genes with homology to any known transporters for inorganic carbon, such as the ABC-type bicarbonate transporter found in Synechococcus PCC7942 , or orf427 in Synechococcus PCC 7002, which has been implicated in CO2 uptake .
The exposure of Prochlorococcus to CO2-limiting conditions and the analysis of the responding genes will help us understand how these processes are performed in Prochlorococcus. It is likely that we will begin to identify completely unknown genes involved in carbon uptake and concentration. These results will have significant implications for understanding C13/C14 isotope discrimination in Prochlorococcus, which in turn has profound implications for calculating global carbon flux and marine productivity.
Organic Carbon: Although we have not demonstrated growth or utilization of organic carbon compounds by Prochlorococcus, analysis of the completed MED4 and MIT9313 genomes has revealed suggestive evidence of genes which may be involved in organic carbon uptake and its potential use as a carbon and energy source. Both strains have genes with closest similarity to known transporters for melibiose and oligopeptides, and possess an intact pentose phosphate shunt, which can allow utilization of reduced organics such as melibiose as a sole source of carbon and energy (i.e. dark, heterotrophic growth). Additionally, both appear to possess an acs homologue, encoding acetyl-CoA synthetase, which is capable of converting acetate (after entering the cell by passive diffusion) into the central metabolism intermediate acetyl-CoA. They have the potential to utilize oxidized organics such as acetate as auxiliary carbon and energy sources by incorporating the carbon into biomass as amino acids and fatty acids, and can derive some energy from an (incomplete) citric acid cycle. However, both strains lack key gluconeogenic capabilities preventing them from using these organic carbon compounds as sole sources for growth and energy. Finally, MIT9313 has a gene cluster whose closest similarity is known transporters for maltodextrins (oligomers of glucose), as well as amylomaltase, the cytoplasmic enzyme that cleaves the oligosaccharide. For many of the catabolic pathways studied in other systems, the nutrient itself can act as an inducer of the genes encoding the transporters and cytoplasmic enzymes. Several well-studies examples include E. coli grown on lactose, maltose, and arabinose .
Temperature – Heat/Cold Shock Proteins
The temperature optimum of Prochlorococcus MED4 for growth is 24° C, and the maximum and minimum are 28° C and 12.5° C respectively . Exposure of cells to temperatures of 28° C results in an immediate decrease in growth rates and is followed by a cessation of cell division and a rapid decline in chlorophyll concentration per cell (Ting. et al, in prep.). In general, the exposure of organisms to sublethal high temperatures results in the selective induction of a specific class of proteins that are highly conserved among archaea, bacteria, plants, and animals . The majority of these heat-induced stress proteins function either as molecular chaperones, promoting the folding of newly synthesized or unfolded proteins, or as proteases, degrading abnormal and misfolded proteins . Past work on protein synthesis patterns of E.coli during steady-state growth near its temperature limits for growth has revealed that the levels of many proteins are increased or reduced . While the levels of several proteins involved in transcription or translation were lower at these temperature extremes, the amounts of those proteins involved in energy metabolism were higher. It would therefore be important to determine whether similar changes in protein profiles are observed for Prochlorococcus. Western blot analyses indicate that the major molecular chaperone, GroEL, is expressed constitutively in Prochlorococcus, as it is detectable both in control and heat-stressed cells (Ting. et al, in prep.). Comparative analyses of the Prochlorococcus MED4 and MIT9313 genomes show that they both possess genes encoding the major molecular chaperones, including groEL, groES, dnaK, dnaJ, grpE, and htpG.
At the other extreme of temperature is cold shock. Exposure of microorganisms to sudden decreases in temperature induces a distinct set of genes, several of which play key roles in countering the effects of cold on membrane fluidity, transcription, and translation . Unlike heat shock, the cold shock genes are not highly conserved among the bacteria, although evolutionary convergence has apparently provided different groups of bacteria with unrelated genes of similar function. Cyanobacterial genes whose expression is induced upon cold shock include the fatty acid desaturase genes desA and desB of Synechococcus sp. , the RNA helicase gene crhC of Anabaena sp. , the heat shock protease gene clpB of Synechococcus sp. , and a family of RNA-binding genes (rbp’s) in Anabaena variabilis . Both MED4 and MIT9313 strains of Prochlorococcus appear to have homologues of desB, as well as two or three other fatty acid desaturases as well, respectively. MED4 has two homologues while MIT9313 has three homologues of the rbp’s of Anabaena variabilis. Both strains have homologues of clpB, and homologues of two cold shock genes of E. coli, the transcriptional terminator gene nusA, and the cold-shock ribosomal factor gene, rbfA . The latter gene’s product alters the ribosome during the transient cold shock, thereby adaptating the translation process to the lower temperature . After this adaptation, expression of the shock genes declines, and translation of the bulk mRNAs of the cell and organismal growth resumes .
Thus, although Prochlorococcus has a very narrow temperature range for growth, it possesses a full complement of genes encoding the putative proteins that have been demonstrated to play a key role in acclimation to temperature stress in other organisms.
Interaction with heterotrophs
In order to model mechanisms of adaptation of Prochlorococcus to simulated variations in environmental parameters it is important to consider the effects of concurrent adaptations of their native co-inhabitants. That is because Prochlorococcus is far from alone in the open oceans (see below, Goal 3), and as such has likely evolved survival strategies that have taken into account the environmental changes that are caused by other members of the native biota. A key nutrient to follow is carbon: under light or inorganic carbon stress, are genes involved in organic carbon uptake and metabolism induced in Prochlorococcus? And if so, how does the presence of heterotrophs affect this response? Are they competitors, or possibly providers of different forms of organic carbon? While it is virtually impossible to re-create the open ocean ecosystem in the laboratory for such analysis, we do have at our disposal several heterotrophs native to these waters that have been co-cultivated with the Prochlorococcus ecotypes. For instance, from the MED4 cultures we have isolated a members of the gamma (Alteromonas alvinellae) and alpha Proteobacteria, and from MIT9313 cultures we have isolated two gamma proteobacteria (Alteromonas macleodii and Halomonas sp. 9313c3) and an alpha proteobacteria (Rhizobium sp. 9313c4) (Bertilsson, unpub). Hence, we have the potential to add back ecosystem diversity to axenic cultures of MED4 and begin to get an idea of the significance that their presence plays in determining the survival responses of Prochlorococcus in the natural setting.
Axenic cultures of MED4 and another Prochlorococcus strain, MIT9312, were found to excrete up to 30% of total organic carbon into the medium during exponential growth, with marginally less in stationary phase (phosphate-limited) cultures (Bertillson and Pullin, unpub). Although there was significant variation between replicates, formic, acetic, glycolic, and lactic acids were detected in the dissolved organic carbon fraction of the cultures (Bertillson and Pullin, unpub). Hence, there is a significant amount of photosynthate excreted into the medium by Prochlorococcus, which can readily account for the maintenance of heterotrophic contaminants. These contaminated cultures thus also represent model systems to study the flow of organic carbon into the heterotrophic population, using the radiolabeling approach proposed for natural populations mentioned below under Goal 3. As laboratory-controlled model systems, we can vary the extent of heterotroph and Prochlorococcus diversity, and the environmental conditions to begin to approximate the importance of diversity and environmental stress affect an ecologically-crucial process of organic carbon flux.
Interactions with Phage
Phage occur at total abundances of 107 ml-1 in the open ocean habitat of Prochlorococcus and are known to outnumber the prokaryotes by a ratio of 10:1 (reviewed in Fuhrman, 1999). Viral infection can have a significant effect on the capacity of autotrophic host cells to fix inorganic carbon (Suttle & Chan, 1993). They also play an important role in regulating phytoplankton population size and dynamics (especially during bloom conditions) and are likely to be one of the forces driving diversity in the natural environment (reviewed in Fuhrman, 1999; Wommack & Colwell, 2000). Viruses also play an important role in the transfer of genetic material from one host to another (Paul, 1999).
A graduate student in the Chisholm lab, Matt Sullivan, has isolated over 50 clonal phage isolates from natural seawater that infect and lyse various Prochlorococcus strains in our culture collection. These include 3 different families of phage from the order Caudovirales (Podoviridae, Myoviridae and Siphoviridae). In addition to the ecological and laboratory characterization of Prochlorococcus cyanophage (which is summarized in Goal 3 of this proposal) it is important to note here that multiple phages, including those from different families, can infect and lyse the same host strain. Furthermore, we have identified putative prophage in our Prochlorococcus MED4 and MIT 9313 genomes and have produced phage resistant Prochlorococcus MED4 strains through prolonged exposure to lysis-causing cyanophage (Sullivan, unpubl.). Resistance to phage can be conferred at numerous levels; mechanisms include mutation of phage receptors, changes in the host machinery required by the phage to produce new phage particles, digestion of the phage DNA by restriction-modification systems and lysogeny (Kruger & Bickle, 1983).
Analysis of global gene regulation
To understand how a cell works one must identify how the repertoire of genes within the cell are regulated at the level of expression. (We use the term gene expression to collectively refer to transcription, translation, and post-synthetic modification of proteins and RNAs) Extensive analysis of global gene expression patterns in other systems, especially E. coli, has revealed a complex circuitry of gene regulatory cascades . Multiple adjacent genes are often co-regulated as operons with a common promoter element. Multiple operons can be regulated by a common regulator, thereby constituting a regulon. Multiple regulons can be regulated by additional regulatory elements, forming modulons. Finally, the stimulon is described as "a group of operons responding to a given environmental stimulus irrespective of a regulatory mechanism" . Therefore, a stimulon may be composed of single or multiple independent regulons. A major goal for the identification of the architecture of a cell would therefore be to identify the stimulons of key environmental perturbations. Tools particularly well-designed for this investigation are the global gene expression technologies, DNA microarrays, which measure the complete transcription profile of the cell (the transcriptome) and mass spectrometry analysis of the cell’s protein profile (the proteome). These technologies also have the capacity to identify possible modulons and regulons by detecting transcriptional regulators whose expression is induced just prior to induction of the modulon / regulon, as described below.
Microarray analysis of the transcriptome
Through genome-wide monitoring of transcription, DNA microarray studies offer the possibility of a genome-wide integrated view of cellular functions . Although transcriptional profiling and physiological state classification have been the central focus of the majority of DNA microarray applications - this technology is also applicable to the investigation of fundamental questions of gene regulation and cell physiology. This is particularly so when one analyzes gene expression patterns at short intervals during the transition from one physiological state to another. Such dynamic profiling enables us to observe the development of a regulatory response, increasing the chances of correctly deciphering cause-effect relationships. This technology is even more powerful when a comparative approach is used, in which we can identify motifs that are conserved in homologous proteins that are likely to be functionally or regulationally important. Studying coordinated gene expression patterns in response to environmental stimuli is also the first step toward interpreting sequence data from novel open reading frames in the genomes by noting their co-regulation under a wide variety of conditions with genes of known function.
The Chisholm Lab has a grant from NSF-Biological Oceanography for the construction of whole genome microarrays for Prochlorococcus MED4 and MIT9313 (with matching funds from her Chair at MIT). The MED4 array is currently in development (see below), and should be available for use soon after the start of this project. The array for the second ecotype, MIT9313, will be developed as the project progresses, but should be available for comparative purposes at about the middle of the project.
Trial Mini Arrays for Prochlorococcus – Progress to date
We have constructed mini-arrays of MED4 to gain experience with the technology and to optimize RNA extraction, labeling and hybridization protocols for our system (see below: Proposed work, Whole Genome Microarrays, for a more detailed description of the microarray protocol). This initial array consists of genes whose transcription patterns in response to environmental stimuli are known from previous experiments with Prochlorococcus as well as a few genes that were expected to respond to each of the perturbations outlined in this study. They contain both highly expressed genes (16S rDNA, psbA, pcbA ), as well as genes known to be expressed at much lower levels such as cpeB and the nitrogen regulatory gene ntcA (Lindell et al. in prep.).
The trial array consists of custom-synthesized 70 bp sense and antisense oligonucleotides (Operon, Alameda, CA) spotted onto Corning CMT-GAP2 slides. Optimizations thus far have led to the ability to detect expression of 39 out of 48 genes (81%) above background noise (by comparing signals at the sense and anti-sense oligo spots; two-sample t test, P < 0.01). The dynamic range for spot detection was over two orders of magnitude. Initial observations of relative spot intensities for different genes on the trial array strongly correlated with the relative expression values obtained from the same RNA sample with an alternative detection method (quantitative reverse transcription-PCR, see below). Future analyses will be performed by determining the intensity ratios of two differentially labeled samples at each spot: the treatment sample and the reference sample. This is to negate potential spot-to-spot printing variations on the microarray that could skew the results and prevent absolute quantitation of RNA (Schena et al. 1995; DeRisi et al. 1997).
Goal 2a: Gene Regulatory Networks in Prochlorococcus
We propose to analyze the responses of Prochlorococcus MED4 to a set of well-defined experimental perturbations (light, temperature, carbon, heterotrophic bacteria, cyanophage) to help us begin to construct a model of their cellular architectures. These parameters have been chosen because of their importance in phototrophy, their importance in understanding connectivity in the microbial community, and because they delineate the vertical and geographical distribution of Prochlorococcus in the oceans. Transcriptional responses will be analyzed using microarray analysis to identify the stimulons associated with each perturbation. Analyses will also be carried out on Prochlorococcus MIT9313 in selective comparisons, following our conviction that a comparative approach will help us begin to assign function to unknown genes, and better understand the regulatory networks in these cells. Our long term goal is to understand the regulatory functions that direct the response of the cell to environmental change ¾ i.e. the mechanisms involved and the cascade of events that bring about acclimation of metabolism to new conditions. This includes identifying the genes whose products are involved in sensing and transmitting messages, the modulons and regulons associated with these regulators, and the mechanism of the phenotypic change associated with these molecular responses.
Toward this end, we will:
2a.i. Analyze the global gene expression patterns of Prochlorococcus MED4 in response to changes in light, temperature, and carbon availability—using whole-genome microarrays and both steady state and dynamic profiling of gene expression;
2a.ii. Do the same for cultures that are exposed to heterotrophic bacteria that we find as significant contaminants in our cultures, and phage that we know to infect Prochlorococcus.
2a.iii. Compare these results with similar, but more selective, experiments done with Prochlorococcus MIT9313 (a low light-adapted strain);
2b. Use informatics to identify potential regulatory motifs upstream of co-regulated genes, as determined from microarray analyses including significant combinations of motifs as we have done for a variety of microbial species.
2c. We will correlate and test the above hypotheses with (i) selection data on mutations in each gene and genetic domain in Goal 3d, (ii) the protein data in Goal 1, and (iii) mass spectrometry of protein complexes selected by solid-phase versions of the motifs.
Environmental Perturbation Experiments – The Raw Material for Analysis
General Considerations for Experimental Design
As discussed above, cell division is tightly synchronous when Prochlorococcus is grown on a light/dark cycle, with growth occurring during the day and division at night . Thus cells grown on light dark cycles have to be harvested at exactly the same time of day for comparisons between conditions, and the results have to be interpreted as cell cycle context dependent. In order to understand the full range of gene expression patterns over the course of the cell cycle/light-dark cycle, our first experiment with gene expression profiles will be performed hourly over a 24 hour light-dark cycle, under conditions in which the population is doubling once per day. We will compare these results with those from an asynchronous culture growing at the same growth rate, to determine how much the asynchrony influences the resolution of the gene expression analyses. These data will provide important information on the cell cycle and light-dependent tasks in the Prochlorococcus cell, as well as inform decisions about the design of future experiments.
To facilitate the growth of Prochlorococcus such that the population grows synchronously and divides once per day, we have modified a standard Percival constant temperature incubator (Braun, unpubl). Whereas a standard incubator regulates light in an all or none manner, our modified system can provide artificial sunlight that simulates a sunrise and sunset. Such a system more closely approximates the light exposure of natural populations, and should avoid unintended shocks of rapid changes in light that can interfere with natural gene expression patterns.
In order to explore the full range and dynamics of gene expression profiles ¾ apart from changes over the light-dark cycle ¾ we have in mind a series of experiments that will examine the transient and steady-state response of Prochlorococcus to exposure to sub-optimal environmental conditions. In these experiments we will be testing both chronic and acute sub-lethal exposure to each environmental variable. We define chronic as steady state growth under sub-optimal growth conditions, and acute as the transition period before the steady state is reached. The chronic sub-lethal exposure experiments will be crucial to our understanding of the cell’s total physiological possibilities within the boundaries for growth (and not just at its optimal growth conditions, which may be rare or absent in nature). Monitoring the transitions into the stress state by the acute exposure experiments may identify the regulatory elements that establish the response to the environmental perturbation. That is, a positive regulator of a stress response may be induced first, after which can be seen its induction of the regulated genes. Therefore, these experiments will involve frequent sampling at short intervals (every 10 minutes for 90 minutes) during the transition period. Another reason for sampling both the transition state and the "acclimated" state is that the gene expression profiles may change after acclimation and resumption of growth, as evidenced by the transitory cold shock response in E. coli . Therefore, perhaps only by looking at the transition period will we be able to identify the genes involved in dealing with the shock of environmental change.
Standard Conditions and Measurements:
Standard growth conditions for each strain will be used as a reference of all experiments with that strain. Both Prochlorococcus strains will be grown in a chemically-defined artificial sea water medium (Zinser, unpubl.), and standard light and temperature levels will be set to those that yield maximum growth rates (mmax), based on our previous work . For all experimental cultures we will measure ancillary parameters such as the concentration of cells when harvested, the growth rate, chlorophyll per cell, and side scatter as measured by flow cytometry (an indicator of cell size) . We will also measure relative DNA/cell using flow cytometric analyses, so that we can characterize where the majority of the cells are in their cell cycles for each sample .
We will grow a series of 5 large volume cultures (10 L) of each strain from which RNA and proteins will be extracted. The variability in transcript and protein expression between these five samples will be determined for each open reading frame and will enable us to determine the reproducibility of the assays and provide a "confidence level" for changes in expression that can be attributed to the environmental perturbations. These samples will then be pooled and used as a reference for all experiments with that strain. This approach will also enable us to test expression levels at 6-12 month intervals to ensure that reference gene expression and protein levels remain constant throughout the course of the project.
Light Shift Experiments
Because light is the easiest environmental parameter to manipulate, and its relatively weak coupling with the chemical environment of the culture over short time scales, this set of experiments forms the heart of this proposal. Since we are just starting gene expression analysis in Prochlorococcus, we are interested in both the steady state expression patterns in cells grown at different light levels, and in the dynamics of expression when the cells are shifted between optimal and sub-optimal levels of light. To this end, we will grow the cells at light intensities yielding maximal growth rate m max, and then shift the intensity either up or down to levels where the steady state growth rate resumes at ¼ m max due to light limitation or photoinhibition. We will also shift them temporarily into complete darkness. Comparative analysis of the global gene expression profiles both in the steady state, and during the transients will allow us to identify key genes involved in photoacclimation and help identify the regulatory elements for this process. By observing the transition from one physiological state to another, and back again, we reduce the chances of observing coincidental regulatory patterns. Temporary exposure of Prochlorococcus to lethal doses of light irradiance and complete light absence may uncover genes essential for adaptation to these stressful conditions.
This type of design facilitates the observation of the regulatory differences between genes activated by all light intensities as opposed to those activated only in response to intense light exposure. Furthermore, it could also reveal those genes that are 1) up-regulated in the dark, 2) regulated proportionally to the input light signal, and even 3) those required only transiently during the state transitions, whose expression levels return to the base line when the new state has been reached. Finally, this design should reveal not only the differences between the three states (static data), but also which genes may be required for transient regulatory phenomena. Cause-effect information can be gleaned from measurements of the time-lag between an input signal and the induction of expression of various genes. Although some regulatory responses will occur on extremely small time-scales, the response of other genes requires the synthesis of new proteins or other interactions before it is observed. Prior experiments with the cyanobacterium Synechocystis PCC6803 suggest that a period of approximately 90 minutes is required for full transcriptional profile development upon transition from dark to light conditions. In the course of this transition, gene expression dynamics vary markedly among various classes of genes . This suggests that for Prochlorococcus, a sampling frequency of 10-20 minutes over the course of a transition will be required. This is a daunting task, but we think it is doable.
Temperature
For the experiments in which we want to look at changes in gene expression in response to temperature, we will grow Prochlorococcus at a temperature yielding maximal growth rate, m max, and then shift it either up or down to levels where the steady state growth rate resumes at ¼ m max maximal growth rate. Analysis of the global gene expression profiles of the three steady state growth conditions will help us define how Prochlorococcus adapts to temperature extremes. For instance, are the levels of proteins involved in transcription or translation lower and the levels of proteins involved in energy metabolism higher at the high temperature extreme, as was found for E. coli ? Analysis of the gene expression profiles of the transition states to sub-lethal and lethal temperatures should help identify the heat shock and cold shock stimulons, and may also identify the regulators of these stimulons. RNA will be extracted from cells following both short (5 min, 30 min, 60 min) and long (24 hrs) exposures.
Carbon
To establish carbon limitation in the growth cultures we will sparge the headspace of a tightly-capped vessel with N2 gas or commercially-prepared air mixtures . The inorganic carbon (primarily HCO3-) in the growth medium acts as a buffer, and by depleting the CO2 from the system, the pH of the medium is expected to decrease. Therefore, the pH will be monitored during these experiments. RNA and protein will be extracted and analyzed in carbon limited and control cultures to identify genes induced during CO2-limitation. Included among this class of genes may be those that increase the carbon concentration capacity of Prochlorococcus, which lacks homologs to known carbonic anhydrases (see Background section). Genes repressed under these conditions may also provide insight into the classes of genes and physiological responses whose function is beneficial only under carbon-replete conditions.
To determine whether exposure to specific organic carbon compounds triggers specific induction of gene expression, we will investigate the RNA and protein profile of cultures exposed to organic carbon under a variety of conditions. Cultures in exponential growth will be monitored in both the light and dark periods of the cycle, as will cultures exposed to prolonged light deprivation (i.e. after cell counts and fluorescence cease to increase). In the latter experiment, cells will be exposed daily to short bursts (10 minutes) of white light (40 mmol m-2 s-1), as such light exposure was found to be necessary for induction of heterotrophic genes and dark growth of the Synechocystis PCC6803 . Carbon compounds to assay will include melibiose, acetate, oligopeptides, and (for MIT9313) maltose and maltotriose (see Background section). Particular attention will be paid to the potential regulatory genes for this heterotrophy: do they exhibit nutrient-specific or more general induction patterns?
Interaction with heterotrophic bacteria
To assess the effects that heterotrophic bacterial populations play in modifying the adaptive responses of Prochlorococcus to environmental stimuli, we plan to repeat some of the above experiments with co-cultures of MED4 and the heterotrophic contaminants found in MED4 cultures and the cultures of other ecotypes. This work will proceed by isolating the heterotrophs on rich broth or minimal media plates and then adding them back to the axenic MED4 cultures. Considerable care will be given to ensure the experiments will be performed with reproducible ratios of the mixes species. Prochlorococcus populations will be monitored by flow cytometry, and the heterotrophs will be monitored by fluorescence in situ hybridization analysis (FISH) with species-specific probes, and by viability counts. Microarray specificity controls will be performed on the heterotrophs in isolation to verify that their RNA will not hybridize to the MED4 array’s oligo spots. We do not anticipate this to be a problem, as several reports indicate that such arrays will not detect RNA’s less than 70-80% identical to the 70-mer oligos (Ward, et al. 2002).
Interaction with cyanophage
As mentioned in the background, phage can have far reaching effects on Prochlorococcus populations. We will initially address the cellular response of Prochlorococcus to infection by phage from the Podovirus family under optimal conditions for growth as well as under select sub-optimal conditions outlined above. We will then address whether infection by a different family of phage (Myoviridae) elicit the same cellular responses from Prochlorococcus. Carbon fixation levels and gene expression (using both whole genome transcriptome and proteome analyses) will be assessed at various time intervals over the 24-48 hour course of the lytic cycle to correlate observed changes with different phases of infection such as adsorption, replication and lysis. We will pay particular attention to four types of genes: those involved in essential cellular functions such as light harvesting and carbon fixation; those that may be induced by the phage such as the genes involved in DNA replication; those that may be involved in Prochlorococcus defense against infection such as restriction-modification systems; and those of the putative prophage. It will be interesting to see whether this putative prophage is involved in conferring resistance to the host when challenged with cyanophage that do not cause host lysis. We will also watch to see if any of the experimental conditions employed induce this putative prophage to a lytic cycle.
We will further assess the effect of cyanophage on Prochlorococcus diversity and evolution by characterizing the interactions between phage isolates and phage resistant host strains. These resistant strains will be tested for resistance to other phage and the mechanisms of resistance will be evaluated at genome, transcriptome and proteome levels. Resistance through lysogeny will be assessed by Southern analysis for incorporation of the phage into the Prochlorococcus genome. We will challenge these resistant Prochlorococcus strains with phage and assess gene expression, at both the transcriptional and translational levels and compare these to the non-resistant strain in order to gain insights into the mode of resistance. Finally, the cost of phage resistance will be assessed through a comparison of carbon fixation in the wild-type and resistant Prochlorococcus strains.
Whole Genome Microarrays – Approach
As mentioned in the Background section, the Chisholm lab has optimized conditions with a trial miniarray, and is in the process of constructing whole genome microarrays for Prochlorococcus MED4 and MIT9313 with funds from NSF. The Prochlorococcus arrays are being fabricated at the MIT Bioinformatics and MicroArray Facility, using a MicroGridII robot (BioRobotics) fitted with Microspot 2500 quill pins and guided by the experience of the facility staff in this procedure. Synthesized oligonucleotides 70 bp in length (Operon, Alameda, CA) will be spotted in triplicate with a 150 µm diameter pin onto Corning CMT-GAP2 slides. A subset of the slides will be tested for quality by hybridization with Cy3 labeled random 9-mer primers (according to Operon protocols). Control spots will include mouse and Escherichia coli genes, with no homology to genes in any of our cyanobacterial genomes, and will be used as both negative (no complementary RNA to be added) and positive (in vitro transcribed RNA of known quantities will be added to sample RNA) controls of our labeling and hybridization.
RNA Isolation, Labeling and Hybridization
Based on our previous work with the miniarray, we expect to use 20 m g total RNA per hybridization experiment which can be obtained from 100-200 ml of an exponentially growing Prochlorococcus culture. RNA will be isolated according to standard protocols (García-Fernández et al. 1998) and enriched for mRNA by removal of rRNA using a commercially available method (Microbe Express, Ambion, Austin, TX). The RNA will be reverse transcribed to cDNA using Amersham’s CyScribe cDNA post-labeling kit and employing random hexamer primers and amino-allyl-dUTP in the nucleotide mix. Cy3 and Cy5 fluorescent labels will be chemically coupled to the amino-allyl-dUTP. Labeled cDNA from both a reference and experimental sample will be combined and hybridized overnight to a printed array at 42 ºC in a formamide solution. After hybridization, slides will be scanned using an arrayWoRx scanner (Applied Precision, Issaquah, WA) consisting of a white light CCD based scanner available at the MIT microarray facility.
Data Analysis from Microarrays
The Applied Precision software that runs the arrayWoRx scanner will be used to apply a best-fit correction transformation for background fluorescence and different fluorescence intensity of the Cy3 and Cy5 dyes. Triplicate spots on each array and experimental replicates will be used to determine the statistical significance of differences in expression to the reference sample. By using the same reference RNA sample for all of the experimental conditions for each strain, expression patterns across all perturbations can be compared. In order to enable efficient data mining, it will be crucial to organize the data in a form that facilitates intercomparisons of the multiple conditions. We have published the first paper on gene expression databases (ExpressDB, Aach et al. 2000*). It covered three types of RNA quantitation (Affymetrix, ratio-microarray, and SAGE) for both bacterial and eukaryotic microorganisms. We can extend this to other microbial species and other functional genomics conditions and and measures (see BIGED database discussion in Aach et al.). We will also assess AMAD (Another MicroArray Database), a freely available (www.microarrays.org) flat file, web driven database system written entirely in PERL and javascript, which provides a means for storage, retrieval, and extraction of microarray data from a centralized web based server. The browser based format will be ideal for managing array data generated both at MIT and by our collaborators elsewhere. We plan to customize our AMAD database so that a multitude of measurable cellular and environmental parameters and experimental details, such as light level, time of day, temperature, growth rate, chlorophyll per cell, media, strain, experimenter, RNA isolation and labeling protocol, and array printing batch are stored for each experiment. This will facilitate more powerful comparisons between all of the perturbations. For example, we will be able to call up all the experiments where the growth rate was ¼ m max, regardless of what was limiting growth, and look at genes induced or repressed under these conditions.
To identify genes that are co-regulated across our experimental conditions we will use a combination of clustering algorithms and motif analysis pioneered by members of this GTL team (e.g. Tavazoie, et al 1999*) as well as cutting edge commercial software packages currently supported by the MIT & HMS array facilities such as Spotfire (Somerville, MA) and Genomax (Informax, Bethesda, MD). Combinations of regulatory motifs as means to achieve the modeling optima in goal 4 would be a computational research focus here too (Pilpel et al. 2001*)
Absolute abundances of RNAs have been determined both using Affymetrix arrays and spotted arrays with genomic DNA as a reference sample (ref J. Bact 183:545-556 and Dudley et al, 2002*). We have developed an increased dynamic range 4-orders of magnitude embodied in our "Masliner" processing software (Dudley et al 2002*).
Expression results for selected genes of interest will be verified by quantitative reverse transcription-PCR. This technique provides a dynamic range of over five orders of magnitude. Quantitation is achieved by detecting the exponential increase in PCR products by fluorescence detection at each PCR cycle, and comparing at which cycle number the amount of products reaches a threshold value. To normalize for RNA extraction, we will either use an externally provided RNA standard or the internal RNA standard, the RNase P gene, rnpB, whose expression is invariant over a light/dark cycle. The Chisholm lab has previously used the rnpB standard to determine the gene expression profiles of several genes over the course of a 14:10 L:D period (Figure 2c, below). The patterns indicate a clear relationship between gene expression and time in the experimental regime, and future work will address if expression is regulated by the cell cycle and/or circadian clock (see above).
Design of unique oligomers
Our group has pioneered the use of full-genome sequence for the design of unique oligonucleotides for arrays for the Affymetrix 25-mers, Operon 70-mers and PCR-based 200-mers (Wright & Church 2002*; Dudley et al 2002*, Selinger et al. 2001*; Badarinaryana et al 2001*). We have another unique advantage in the added precision about actual protein start sites via the proteogenomics software described in goal 1 (Jaffe et al.2002*). We are collaborating under separate funding with a group in Houston (Linxaio Gao) and one in Boston Univ. (Rostem Irani) on micromirror oligo array synthesizers (Singh-Gasson et a. 1999). Because these technologies are currently well-suited for the inexpensive synthesis of a limited number of arrays containing a large number of oligos, we will use these for prescreening large numbers of oligonucleotides for hybridization with genomic DNA to empirically pick the best oligos for Affymetrix makes masks. Evidence that genomic DNA is well correlated with RNA in oligo utility (Selinger et al. 2000*) supports this strategy. The highest resolution DNA-protein crosslinking (aka "location", see goal 2c) experiments and fine-structure whole-genome mutant selections would merit the precision of the highest density arrays (500,000 oligonucleotide 25-mers). Many experiments such as initial surveys of fine time series would be feasible with as few as 2000 oligos (one per gene), which would be considerably less expensive when done in an array-of-arrays format. For these very small subsets even stricter criteria for quality of oligo choice is critical since so much rides on single oligos.
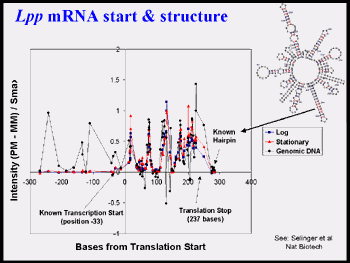
Figure 2d, above, emphasizes the variation in signal with oligos selected by an early algorithm and the utility of using genomic DNA controls for RNA experiments (see Selinger et al. 2000* attached).
Goal 2b: Use informatics to identify potential regulatory motifs
upstream of co-regulated genes, as determined from microarray analyses including significant combinations of motifs. We have done this for about 20 microbial species (Mcguire & Church 2000; Mcguire et al. 2000; Hughes et al. 2000; Pilpel et al. 2001; Zhu, et al. 2002). We will look for correlations with operon structure and conservation of location in microbial chromsomes (Cohen et al. 2001*) especially in light of possible insights available from goal 4e (4D-cell model). The major challenge for the small genomes is paucity of examples of a given motif. This can partially rectified through the use of comparative DNA (motifs from multiple related genomes) and the location data (see below). Once the associations among most of the key proteins and motifs is established (including possible competition and cooperations), then various surrogate measures can be used to measure the occupancy of each site for example methylation protection (Tavazoie and Church 1998*)Goal 2c: We will correlate and test the above hypotheses
with (i) selection data on mutations in each gene and genetic domains in Goal 3d, (ii) the protein data in Goal 1, (iii) location data and (iv) mass spectrometry of protein complexes selected by solid-phase versions of the motifs. One might naively expect some correlation among each of these four sets. However, it is the inevitable rich set of exceptions and combinations that makes for increasingly accurate biosystems models.Location data refers to the antibody selection of DNA-protein complexes crosslinked in vivo by formaldehyde (or other agents, see goal 1c). For the location data we will use the same protocols that we have applied to Caulobacter (Laub et al. 2002*, see attached) on the other genera. The antibodies will be raised against the most abundant putative DNA binding proteins based on goal 1a. One obviously valuable data set will be based on antibody to the RNA polymerase. This will determine the location of paused and elongating molecules during each of the time-series. This will be done with and without initiation inhibitors as we have done in E. coli to establish elongation sites and decay rates. In addition to helping to dissect the chain of events leading to regulated level of various RNAs, these data provide anchoring points for potential associations of the nascent protein chains in the goal 4e models.
The solid-phase double-stranded DNA selections for proteins or protein complexes present in cell extracts will be based on a liberal set of motifs derived in goal 2b. The methods will be analogous to those in Bulyk et al. 2001*, but will depend more heavily on the ability of many such selections to act as controls for one another. Some proteins or complexes will have a high non-specific binding and will turn up in all of the many of the selections. We will determine the reproducibility of the assays as a function of other proteins in the extracts and will calibrate the quantitation using ds-DNA and protein complexes which we previously characterized (.e.g. transcription factor EGR1).