"Goal 3 -- Characterize the Functional Repertoire of Complex Microbial Communities in their Natural Environments at the Molecular Level."
Goal 3 a and b: Background and Progress to date
Understanding the nature of diversity and of functional units in microbial communities is one of the major challenges in microbiology, ecology and evolutionary theory. Although ribosomal RNA approaches have provided first steps towards diversity estimation, and are widely used as a proxy for unique bacterial ‘types’ in natural populations, it remains unknown at what level of genetic resolution an ecologically functional unit must be defined. Furthermore, although genomic studies on cultivated bacteria have resulted in important and unexpected insights into the processes and patterns of genome evolution, it remains unclear how these insights may be extended to populations that co-occur in natural environments. Many crucial questions, such as at what level of structural similarity genome evolution is driven by homogenizing versus differentiating mechanisms, can only be answered by analysis of co-occurring genomes at different level of phylogenetic relationships.
Goal 3a
We will use the cyanobacterium Prochlorococcus as our central model to explore in detail the genomic variation that occupies a single dominant and well-defined niche in the ocean. This will be accomplished by flow sorting the Prochlorococcus cells away from the rest of the microbial community, constructing a BAC library, and, depending of the diversity encountered, either assembling the complete genomes or large contigs to determine the structure of co-existing genomes. Should assembly of large genome portions not be possible, we will provide anchors for the bioinformatic/evolutionary analysis by identification homologous genes/genome regions in the BAC libraries (see below). We will also measure the diversity of co-existing Prochlorococcus in the four samples by rarefaction of a number of different gene markers and by application of in situ amplification techniques.
Furthermore, we will estimate the overall diversity and nature of phylogenetic and functional variation in genomes of uncultured bacterioplankton co-existing with Prochlorococcus. This is to delineate the diversity of the total bacterial community – a task that has remained elusive yet is crucial for effective implementation of environmental genomics. We have recently discovered through elimination of a major artifact that bacterial diversity in the coastal ocean has likely been overestimated by at least an order of magnitude. We seek to extend this approach to the open ocean systems, and complement diversity estimation by capturing and assembling large genome fragments of important members of the bacterial community. This will provide estimates of the extent and nature of genome variation on a community level. We will also assess to what extent function is conserved in bacterial communities under different environmental regimes by development of ‘functional genotype multiplexing’ through an extension of ‘in situ amplification’ protocols developed by the Church lab. These will allow the simultaneous identification of phylogenetic identity and presence of functionally relevant genes in the genomes of uncultured prokaryotes.
For both tasks under goal 3a, we will use bioinformatics and evolutionary analysis to assess the nature of the diversification process. That is: (1) Survey genes representing different functional categories (informational, central metabolic, photosynthetic, catabolic, etc.) for their prevalence and sequence diversity; (2) Distinguish purifying selection (maintenance of function) from function change (or loss) by comparisons of DNA versus protein divergence (synonymous vs. nonsynonymous sequence changes); and (3) Look for evidence of recombination and gene transfer through congruency of phylogenetic trees of genes, unusual codon usage, and local gene order; and (4) identify potential prophage inserted within the Prochlorococcus genomes to characterize the relationships between Prochlorococcus and prophage diversity.
Goal 3b
We will explore the functional connection between the dominant autotroph Prochlorococcus and co-existing heterotrophic bacteria. Our goal here is to determine the extent of specific cell-to-cell interaction in the well-mixed oceanic environments. We will determine whether specific carbon compounds known to be excreted by Prochlorococcus are taken up by specific heterotrophic bacterial populations indicating selection for species networks or whether carbon transfer is guided by chance encounters of individual cells. We will combine DNA microarray and radiotracer techniques in a novel application, the ‘functional diversity array’, which will allow us to identify and link carbon sources and sinks within the microbial community. The FDA will be complemented by a new technique, which we term here ‘single cell activity multiplexing. It combines in situ amplification from single cells in acrylamide matrix with quantification of uptake of radiotracers.
Field Dynamics
The seasonal dynamics of Prochlorococcus populations have been well documented in the N. Atlantic and Pacific (from the USJGOFS HOT and BATS Time series stations), and in the Gulf of Aqaba in the Red Sea. ¾ the three sites we have chosen for constructing the BAC libraries. These data-sets have information on the total Prochlorococcus "meta-population" ¾ i.e. all of the cells that are identified as Prochlorococcus based on their light scatter and fluorescence signature using flow cytometry. This includes all of the ecotypic diversity at a particular site, and thus describes the outer bounds of the collective niche of this group.
The dynamics of the meta-population are distinctly different at the three sites, thus providing us with different selection regimes for our field studies. At the HOT site in the Pacific there is very little seasonal change; the surface mixed layer never extends below the euphotic zone, thus nitrogen remains undetectable in the surface mixed layer throughout the year . Prochlorococcus are fairly uniformly distributed above and below the mixed layer year-round at this site. At the BATS site in the Atlantic, the water column is stratified in summer, with a 20m mixed layer, but mixes down to about 200 meters during the winter. Prochlorococcus abundance is low in the mixed layer in summer, and very high in the static sub-surface chlorophyll maximum layer at the base of the euphotic zone. In winter it is uniformly distributed throughout the mixed layer, in moderate abundances . In the Gulf of Aqaba of the Red Sea, the scenario is the most extreme. Here the deep waters are never cold enough to sustain strong stratification, thus in winter the water column mixes down to at least 600m and the Prochlorococcus population is undetectable. As deep mixing subsides in April, the population re-emerges and by July there is a huge sub-surface maximum at about 100 meters, with cell densities as high as 106 cells ml-1. This is accompanied by smaller population in the shallow surface mixed layer .
Thus these three sites provide us with very different selective regimes for the Prochlorococcus meta-population. One extreme is the situation in the Red Sea where a very large population is built up from an extremely small founder population after winter mixing. This population is established below the mixed layer, where low light conditions are relatively stable, and persists until the onset of deep winter mixing. The other extreme is the surface mixed layer at the HOT site, which does not undergo much seasonal perturbation, but experiences short term light fluctuations in the mixed layer throughout the year. In the middle is the BATS site, where moderate seasonal forcing exists. With this in mind, we will strategically select depths and seasons for sampling among these three sites for the construction of our BAC libraries.
Ecotypic Diversity in Prochlorococcus
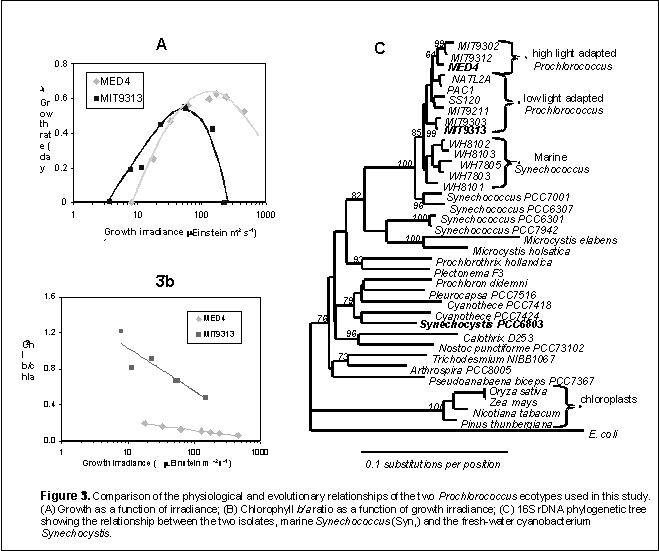
The Chisholm Lab has isolated 55 strains of Prochlorococcus into culture from diverse oceans. Phylogenies constructed using rDNA sequences from a subset of the collection reveal clades that cluster into ecotypes (Fig. 3C, below) according to their optimum and minimum light intensity for growth, and the range of their chl b/a ratios (Fig 3A, 3b). . The ecotypes differ at the 16S rDNA locus by about 2% (Fig. 3C) and the rDNA sequence variability among our cultured isolates can be directly related to that observed in the field . More refined analysis of phylogenetic relationships among isolates based on the 23S and ITS regions of the rDNA locus support the distinction between the two types. High-light (HL) adapted isolates are closely related and cluster together in a shallow clade, while the low light (LL) adapted isolates are more divergent . Ecotypes have also been shown to be distinct in terms of their optical properties, and the structure/composition of their photosynthetic apparatus as well as in their Cu tolerance and Co requirement .
We have also shown that the HL ecotypes can only use ammonia as a nitrogen source, while the LL ecotypes can utilize both ammonia and nitrite . In contrast to their close relative Synechococcus, none of the Prochlorococcus isolates can use nitrate. These physiological observations were confirmed by whole genome analyses: The HL strain MED4 lacks the genetic machinery to reduce nitrate or nitrite, whereas the LL strains do contain the genes for nitrite reduction (see below). Thus we can begin to connect genome diversity with niche diversity: The ecotypes that thrive at high light (i.e. surface waters) have lost the machinery to use oxidized forms of nitrogen, which is consistent with the predominance of regenerated ammonium in surface waters. In contrast, those that thrive in low light have retained the ability to use nitrite, which is usually relatively abundant at the base of the euphotic zone. Depth distributions of ecotypes in the field are consistent with the HL and LL designation .
There is no relationship between the phylogenetic affinities of the different cultured ecotypes (Fig. 1C) and their ocean of origin . This is consistent with the observation that a 2% 16S rDNA sequence difference between the ecotypes translates into separate evolutionary history of, very roughly, 100 million years , whereas the mean global circulation time of the oceans is on the order of thousands of years . That is, microbial distribution in the oceans is determined by ecology, not by geography, per se.
Thus we hypothesize that multiple ecotypes of Prochlorococcus co-exist in all oceanic environments, alternating in dominance along spatial and temporal gradients. These ecotypes are descendants of a common ancestor yet have been shaped by evolutionary mechanisms that lead to diversification. Much of this diversification is gradual along clonal lineages, as evidenced by the rRNAs; however, major change can be introduced by gene loss and rearrangement, or lateral gene transfers as evidenced by comparison of two Prochlorococcus genomes (see below). Ultimately, ecotypes arise that are genomic hybrids, consisting of families of genes whose co-occurrence has been selected for based on the probability of co-occurrence of particular environmental conditions in the oceans. One of the goals of this proposal is to begin to understand the full extent of this diversity – from gradual changes to major genome differences - and, ultimately, its relationship to the dynamics of the environment.
Comparative Genomics of two Prochlorococcus Ecotypes
The DOE’s Joint Genome Institute has sequenced the genomes of Prochlorococcus MED4 and MIT9313. MED4 belongs to the more recently evolved HL clade of Prochlorococcus, while MIT9313 belongs to the LL clade (Fig. 3a). Over the course of this differentiation there has been a dramatic reduction in genome size (Table 1). MED4 has the smallest genome for any known oxygenic phototroph, with 1.7 Mbp and approximately 1700 potential genes (Table 1). A comparison of the genomes of these two ecotypes reveals a common core of ca. 1300 genes, and a large group of genes, conserved in both genomes, are of unknown function. In addition, each genome contains a significant number (200-600) of genes that are (currently) unique (Table 3a) ¾ the majority of which (about 60%) are of unknown function. Alignment of the two genomes demonstrates that they are mosaics of blocks of genes with significant rearrangement (Fig. 3b), and closer inspection reveals that even between conserved blocks, insertion/deletion events have led to further differentiation (see below).
Concurrent with the reduction in genome size in MED4 is a dramatic reduction in %GC content, leading to different codon and amino acid usage patterns compared to MIT9313, and a reduced number of genes encoding regulatory proteins (Table 1). For example the MED4 genome contains 4 histidine kinase motifs (Tolonen unpubl. data) in comparison to the 43 found in Synechocystis PCC6803, a related fresh water species. In fact, of all the genome sequences available on the Integrated Genetics Website, MED4 has the fewest histidine kinase motifs, implying that it has very few regulatory circuits and networks. Superficially, this might suggest that energy is not limiting in the high light environment, so that a small core of constitutively expressed biosynthetic pathways is emphasized over a broader set of regulated assimilatory pathways.
Ecotypic Differences at Selected Loci.
The comparative study of laboratory isolates of Prochlorococcus with regard to detailed features at selected loci (selected either for their universal function of their ecological relevance for this particular organism) has begun to yield some insights into the genetic basis of ecotypic diversity. We do not have room to review all that has been unveiled thus far, but comparisons of the photosynthetic apparati of the two ecotypes can be found in two of our recently published review articles (see ), one of which can be viewed at http://web.mit.edu/chisholm/www/prog.pdf.
One particular comparison is compelling with regard to the importance of deletion events in the evolution of ecotypes. As mentioned above, Prochlorococcus is unusual in that it cannot utilize nitrate as a nitrogen source , and only the LL ecotypes can utilize nitrite. The HL ecotypes are limited to ammonium and urea as their nitrogen sources, which is consistent with their predominance in surface waters where these regenerated forms of N dominate. Prochlorococcus’ close relative Synechococcus, however, can utilize all three forms of N.
Comparative genomics has revealed that this makes sense when you consider the evolutionary origins of these three ecotypes as well as the ecological niches they now occupy (Fig. 3c).
Serial deletions of segments of the N-metabolism regulon, have resulted in the sequential loss of the nitrate and then the nitrite reductase genes as the LL Prochlorococcus ecotypes evolved from Synechococcus, and the HL ecotype evolved from its LL relative (Post et al, unpubl.). The net result is that the HL ecotype dominates high-light surface waters where ammonium is the dominant N source and the LL ecotype dominates deeper waters where light is scarce but nitrite is often abundant. Their close relative and Synechococcus has a very broad niche with respect to N utilization, and thus is capable of bloom formation when NO3- upwells from the deep water. Synechococcus cannot, however, grow at the very low light intensities at which LL Prochlorococcus thrives. Thus these deletion events have played an important role in niche diversification among these ecotypes. It is likely that as we begin to compare other genes that differ among the ecotypes we will gain clues as to other environmental "drivers" for this diversification. Indeed, similar deletion events can be seen in the photosynthetic apparatus of .
Prochlorococcus cyanophage
Almost every Prochlorococcus isolate in our collection has shown susceptibility to lysis by naturally-occurring cyanophage (Sullivan, unpubl). Several phage have been cloned, and their host ranges have been found to vary considerably. Some phage are capable of infecting only a single host while others infect multiple hosts even spanning both ecotypes of Prochlorococcus and in some cases a second genus of marine cyanobacteria, Synechococcus.
In addition to lytic phage, prophage have recently been shown to exist in natural marine Synechococcus communities . Using bioinformatic approaches to detect possible prophage in our Prochlorococcus genomes , we have detected possible prophage present within the MED4 (~35 kb+ in size) and MIT9313 (~ 20+ kb in size) genomes (Sullivan, unpub). A key objective for future work will be to determine if these represent functional phage capable of being induced to a lytic stage, or are remnants of inactive phage. As we search for novel means of creating a working genetic system, the benefit of a functional prophage might prove invaluable for future genetic manipulation in Prochlorococcus.
IMPORTANT NOTE:
Polz and Chisholm recently submitted a NSF Biocomplexity proposal (along with Hiroaki Shizuya and Gary Olsen) to do the BAC Library and fingerprinting work described herein for Prochlorococcus at the three study sites. That proposal did not include analysis of the rest of the microbial community ¾ or the connectivity between it and Prochlorococcus ¾ that we are proposing here. If the NSF proposal is funded, it would support the construction of a minimum of four BAC libraries from flow-sorted Prochlorococcus cells obtained from the Bermuda Atlantic Time Series Station (BATS), and the Hawaii Ocean Time Series Station (HOT) and the Gulf of Aqaba in the Red Sea. One of these libraries would be fingerprinted to determine contigs of the co-existing environmental populations, while the others would serve as reference libraries. The NSF grant only includes funds for sequencing of selected genes of environmental relevance but not of whole genomes. Thus, we propose full sequencing of the fingerprinted library by the JGI under the auspices of this grant (see estimate for coverage and cost estimate). Should the NSF Biocomplexity not be funded, we would ask the JGI to carry out both BAC library construction and sequencing of the environmental Prochlorococcus BAC library.
Measurement of Genomic Diversity of Natural Communities
Diversity is a central ecosystem parameter as a measure of co-existing, interacting and co-evolving genomes. Although we have, in principle, learnt how to measure bacterial community diversity via measurement of ribosomal RNA diversity, reliable estimates are still limited to simple environments. In fact, a recent review showed that no complex marine environment has been sampled sufficiently and so bacterial diversity remains an open question . Molecular diversity studies typically circumvent culture of organisms by directly collecting cells from the environment, extracting mixed DNA, and PCR amplification and cloning of variants of specific homologous genes . Ribosomal RNA (rRNA) genes are particularly useful because they allow universal phylogenetic differentiation of organisms, and the rRNAs themselves provide excellent targets for identification/quantification of populations via in situ or slot blot hybridization . Furthermore, because in many bacteria, rRNA content is positively correlated to growth rate quantification of specific rRNA in natural samples can give information about the relative activity of populations . However, we have recently discovered that although this lack of diversity estimates is in part rooted in technical difficulties, more importantly, methodological problems may lead to an explosive accumulation of sequence artifacts (Thompson et al. 2002). The discovery of this methodological problem has recently enabled us to estimate the total ribotype diversity in a coastal bacterioplankton community (see preliminary results).
This lack of data on ribotype diversity is compounded by an absence of information on genomic variation that may lead to functional variation within ribotypes (genomes with identical rRNA sequences) that co-occur in the environment. Thus, the functional unit represented by diversity measurements can currently not be ascertained. Only a single study analyzed an environmental BAC library constructed from a sample of coastal bacterioplankton. They detected two archaeal clones belonging to a single ribotype and several clones with closely related ribotypes. Analysis of genes that flank the rRNA operon revealed that homologous genes were present but that there was sequence variation in all clones. However, in the clones with identical ribotype the variation was limited to synonymous substitutions indicating functional equivalence . While this suggests that there is indeed genomic variation within ribotypes, more extensive studies are obviously needed to improve our understanding of overall genome variation, especially as it relates to ribotype variation and the relationship of ribotype diversity to ecotype. In addition, sequence variation contains valuable information about the mechanisms and history of the forces that structure environmental populations and genomes.
Mechanisms of diversification and selection
In an ecological context, microbial diversity will ultimately be determined by the rates and mechanisms that generate genetic change and the degree to which such changes are removed through selection and drift. In the extreme, diversity could be manifest as a virtually immeasurable continuum of sequence and genome variants. Alternatively, and more likely, biotic and physical factors in the environment may regularly purge variation from natural communities, leading to discontinuous and limited genomic variation. Several mechanisms that may introduce change into bacterial genomes have been inferred from experimental studies and comparative sequencing. These include clonal diversification (accumulation of point mutations that are passed vertically along lineages), gene loss, intragenomic rearrangements, and horizontal mechanisms like recombination and lateral gene transfer. Additionally, insertion sequence (IS) elements, transposons and phages may play an important role in diversification of genomes. Of these, clonal diversification and recombination will introduce change into existing genes without altering overall genome structure while all other mechanisms will change gene order or content.
Though point mutation is the ultimate source of sequence change, other mechanisms acting in concert may considerably modify its effects. Aside from the generation of evolutionary novelty, clonal divergence may eventually lead to isolation of populations from recombination, a consequence that may be of equal or greater importance . Recombination rates have been shown to decrease exponentially with sequence divergence in Bacillus, Streptococcus and Escherichia . For example, in a study comparing nucleotide divergence in the rpoB gene to transformation frequency in Streptococcus, transformation became increasingly rare as gene sequences diverged and was no longer detectable at 27% difference. Such genetic isolation, reinforced and modified by ecological factors, such as geographic isolation, population effects and selection, may ultimately lead to the accumulation of functional differences. Thus, the degree to which sequence diversity is continuous or discontinuous within and among clonal populations may have considerable ecological significance.
While once considered to lead primarily to homogenization on the population level, recombination can enhance genetic diversity when it occurs between clones in a structured population . In the classical sense, recombination allows the co-existence of polymorphism and so expands the potential niche of a species. However, due to its dependence on genetic similarity, it is difficult to predict recombination rates between populations in the absence of sequence information for co-occurring populations. Rate estimates have been obtained for E. coli isolates from the ECOR collection, suggesting that sequence divergence due to recombination is 50-fold higher than that due to mutation . In the extreme case of H. pylori, which occupies a niche in absence of competitors, appears to be panmictic . Explicit tests for estimating recombination are now available but to date these have only been done for pure-cultured isolates. One of our explicit goals is to apply such tests to genomes in naturally occurring communities.
Lateral gene transfer has also undoubtedly played an important role in bacterial evolution . Well known examples include the pathogenicity islands in several bacteria which can be traced to phylogenetically distinct groups . The rates of transfer in the environment are unknown, but may be enhanced if the genomes contain regions that are predisposed to accept foreign genes. For example, recently described transposons harbor integrons that target specific sites in the genome that can integrate and express open reading frames . They appear to be widespread, can be present in multiple copies in genomes, and have been found associated with resistance genes. However, although integron mediated lateral gene transfer may be one of the major factors that introduce variation into bacterial genomes, at the current state of environmental genomics, its effect may be difficult to estimate as it acts in narrowly circumscribed islands within the genome.
Genome rearrangements and gene loss may also have significant effect on structuring genomes, however the importance of these processes in the environment is unknown at present. It is likely that most such events are detrimental in genomes that have long co-evolved with their environment and so may rarely be detected among closely related bacteria in natural environments. Nonetheless such events may be more favored under conditions of rapid environmental change, such as transfer to a culture medium, and so may be more frequently represented in existing databases (which are dominated by cultivated organisms) than in naturally occurring genomes. For example, even genome disruption by IS elements may be relatively rare in environmental populations as suggested by a recent comparison of Yersinia pestis strains which showed identical IS element numbers and locations in all strains of the biovar responsible for the plague pandemic in modern times even though these IS elements integrate at random locations into the genome . Ultimately, however, the impact on naturally occurring genomes by gene rearrangements, gene loss, IS elements and more targeted insertions such as integrons will likely have to await environmental genomics approaches capable of examining large numbers of whole genomes or large contiguous genome fragments. We believe that the approach proposed here, will allow exploration of several significant features of genome diversity and inference of mechanisms of genome evolution under different environmental regimes.
Diversity and Culturability of Bacterioplankton
As outlined above, the majority of bacteria in the environment have remained uncultured. This also applies for bacterioplankton species. This has largely been determined by comparison of results from isolation attempts, direct counts of cells, and, during the last decade, molecular approaches . For marine bacterioplankton communities, culture-independent approaches have lead to several important generalizations . First and foremost, it is believed that culture approaches, which isolated of bacteria on media with high substrate concentration, have lead to isolates that poorly represent the dominant rDNA sequences recovered. Thus, it has become customary to classify marine bacteria into culturable and unculturable . Only alpha-Proteobacteria of the Roseobacter clade, Cytophaga/Flavobacerium representatives and cyanobacteria generally are both recovered at high frequency in culture collections and in clone libraries. Other common isolates, particularly some gamma-Proteobacteria genera (e.g., Vibrio) grow on marine agars but occur infrequently in clone libraries. Among the groups that have evaded cultivation to date are the SAR11, SAR116 and SAR86 clusters and the Actinobacteria. These are frequently dominant in clone libraries and appear to be cosmopolitan judging from their occurrence in clone libraries from a variety of habitats.
Despite great progress in understanding of bacterioplankton diversity, major questions remain. First, we still do not have good estimates of total diversity in bacterioplankton communities. Second, dynamics of plankton communities using clone libraries has only been addressed infrequently. Both are problems of insufficient sampling of clone libraries ; however, this can now be addressed by using equipment increasingly available through genome centers. Third, the ecological role of the uncultured bacterial phylotypes is unknown; however, as detailed below exciting new approaches will allow significant progress.
New Approaches to determine structure-function relationships
An exciting recent extension of molecular approaches is the simultaneous determination of structure (phylogeny) and function (metabolism) of microbial populations. Environmental samples are amended with isotopically heavy substrate, which is metabolized by the community. In one set of methods, active populations are identified by incorporation of 13C from the added substrate into biomass and subsequent detection of population specific tracer molecules such as DNA or polar lipid derived fatty acids . A second method combines in situ hybridization by phylogenetic oligonucleotide probes together with 14C based autoradiography, allowing simultaneous determination of activity and identity .
We are currently developing a conceptually similar but more broadly applicable approach, the Functional Diversity Microarray (FDA). This combines isotopic labeling of active populations with measurement of population diversity using DNA microarrays.
Overview Goals 3 and b
We propose to analyze the microbial community from three oceanic environments with disturbance regimes that vary over different time-scales (daily, months, seasonal). We will focus on Prochlorococcus, which is the dominant primary producer in these environments, and its functional connection to the bacterial community. We will explore the nature of genomic variation and modes of diversification within the single environmental niche occupied by Prochlorococcus. We will further determine the extent and nature of variation of bacterial ribotypes co-existing with Prochlorococcus under the different environmental regimes. Finally, we will explore connectivity between Prochlorococcus and heterotrophic bacterial populations by determining the patterns of carbon transfer between this dominant primary producer and co-existing heterotrophs.
We have chosen the specific environmental sites below to maximize differences in selective regimes both with regard to seasonal disturbance, and short-term mixing dynamics (see background section):
(1) HOT – Summer surface mixed layer: A population which has been isolated in the mixed layer for most of the year, experiencing fluctuating high light/low nutrient environment (minimum disturbance with short term fluctuations)
(2) HOT – Summer, below the mixed layer: A population that has been isolated from the mixed layer for most of the year and experiencing relatively constant low light/low nutrient environment (minimum disturbance – long-term stability)
(3) BATS – Summer deep chlorophyll maximum layer: A population that has been isolated from the mixed layer for several months and experiences a relatively constant low light environment, and relatively higher nutrients (Intermediate disturbance – short term stability).
(4) Red Sea – Summer deep chlorophyll maximum layer: A population experiences relatively constant low light and exists only June – Sept, before it is essentially eliminated by deep winter mixing (maximum disturbance – short term stability)
One of these libraries ¾ to be determined from the analysis of gene diversity described below ¾ will serve as our ‘reference library’ and will be assembled into contigs by fingerprinting (see note on matching funds from NSF). This library will also be targeted for potential full sequencing under the auspices of this proposal Many of the questions posed will be addressed using this reference library, and this will represent phase I of our work. In phase II, we will move into the comparative stage where we compare loci and genes in the other BAC libraries.
Specific Questions
What type and extent of genomic variation exists in co-occurring Prochlorococcus populations?
We will determine what common genomic backbone and superimposed variation exists in the genomes of co-occurring Prochlorococcus. We will initially approach this by fingerprinting the entire BAC library from the environmental location we have found to display highest number of sequence variants in the diversity screening. Depending on the genomic variation encountered in the sample, the fingerprinting will provide us either with completely assembled genomes or with large contiguous portions of the genomes (at a minimum the average
Size of a BAC clone). We will completely sequence large regions of the genomes (or contigs) anchored by informational genes and pathways identified largely from the two sequenced ecotypes. This will provide us with a rich comparative dataset and will form the foundation for comparative analysis of the BAC libraries from the different environments.
What are the major modes of diversification of these Prochlorococcus populations?
We will analyze the gene sequences and genome architecture we encounter in the completely fingerprinted and in the partially characterized BAC libraries for quantitative and qualitative information on mechanisms that drive the evolution of these genomes. We will identify contigs containing rRNA operons and target these for complete sequencing. We will group the contigs by rRNA similarity and analyze the sequences for quantitative evidence of importance of (point) mutation vs. recombination and qualitative evidence for differences in overall architecture. The first will be done by identification of at least 6 orthologous genes that are 10s of kbp apart on the contigs and comparison of their DNA and protein sequence divergence and congruence of phylogeny. The second will be approached by contrasting the contigs for differences in gene arrangement, duplication, gain and loss. Beyond presence and absence of genes (or, more generally, genome regions), relative divergence of genes, synonymous vs. nonsynonymous changes, strength of codon bias, and unusual ("alien") codon usage will also be examined. We will strive to include genes with demonstrated differences in expression level and those with markedly different numbers of interactions within the cell.
What is the genomic diversity in key genes and pathways that are under different selection regimes?
We will determine to what extent the differences in environmental disturbance regimes transcend to diversity on the genome level. An important question is whether the two extremes, high stability (HOT) and population crashes (Red Sea) lead to reduced diversity as opposed to the intermediate disturbance regime (BATS). We will address this by comparing evidence of overall diversity in the marker genes obtained by PCR and in specific genes and pathways from the BAC libraries. Initially, we will concentrate on genes that have already been shown to be important in determining ecological success of Prochlorococcus (see Background) but important additional genes are likely to be identified through the ongoing development of Prochlorococcus DNA microarrays in the Chisholm lab. We will identify BAC clones carrying target genes by hybridization with gene probes constructed by PCR and determine sequence diversity. The comparison of genes under strong (e.g., transporters, light-harvesting apparatus, N and P uptake) and weak (e.g., informational, central metabolism and housekeeping genes) environmental selection will help identify key differences.
How closely do cultured Prochlorococcus isolates resemble environmental genomes, and what types are most readily isolated from the environment?
Prochlorococcus is one of the few ecologically dominant microbes for which an extensive culture collection exists. Thus, we will determine in an exemplary fashion how well the diversity among the cultured strains represents the environmental diversity. This will be accomplished by comparing sequence diversity in some of the same key loci used for the above two questions. In cases where genes can be associated with specific isolates, or at least linked with a common organism through the BAC assemblies, phylogenetic trees will be constructed and compared for consistency among genes.
How many bacterial ribotypes co-exist under the different environmental selection regimes and what is the nature of their genomic variation?
We will compare sequence diversity in 16S and 23S rDNA clone libraries obtained from the three different environmental selection regimes. These genes are the standard in diversity work and estimates of their total diversity and overlap in distribution is needed as a first step for future environmental genomics applications. We have recently shown that rarefaction of such libraries is possible by a combination of high-throughput technology, new statistical methods, and by modification of existing PCR amplification schemes that avoid generation of artifactual sequence diversity (see background). Furthermore, we will adapt the ‘in situ amplification and sequencing’ technology developed by the Church lab for rapid determination of overall ribotype diversity in the environment.
What is the relationship between structure (phylogeny) and function in the bacterial communities from the different environmental regimes?
We will expand this question from the detailed exploration of diversity within the Prochlorococcus populations to the co-existing uncultured bacterial community by two new approaches. First, we will use our newly developed ‘capture and walk’ technique that allows us to use oligonucleotide probes specific for a ribotype to pull large genome fragments (up to 20 kb) from the environment. These can be cloned and sequenced, and probes complementary to their ends can be designed for capture of contiguous fragments. Thus, clone libraries that are samples of the co-existing diversity within identical and similar ribotypes can be assembled and the diversity of associated genes explored. Second, we will adapt the ‘in situ amplification’ technology to a functional multiplexing in which co-localization of specific structural and phylogenetic marker genes can be identified in a high throughput manner. We will concentrate on uncultured bacterial ribotypes found to be either numerically dominant or to be an important link in carbon transfer from Prochlorococcus to the bacterial community (see below).
What are the patterns of functional connections between the dominating autotroph Prochlorococcus and the heterotrophic bacterial community?
We will explore to what extent carbon compounds excreted by Prochlorococcus structure the heterotrophic bacterial community by application of our ‘functional diversity array’ (FDA). This allows simultaneous identification of microbial ribotypes and determination of growth on specific carbon substrates. The rRNA clone libraries from the different environments will be used as templates for construction of ribotype specific oligonucleotide probe arrays. These arrays will be hybridized against total rRNA from samples incubated with 14C-labeled carbon substrates, which were collected from Prochlorococcus or identified as important exudates. Populations, which actively metabolize these substrates can be identified by the radiolabel accumulated in their rRNA allowing qualitative assessment whether carbon transfer routes are dictated by chance encounters between heterotrophic and autotrophic populations or whether specific associations may have (co)-evolved over time.
What are the relationships between Prochlorococcus and prophage diversity?
We will use bioinformatics approaches to identify candidate prophage in the BAC library clones of Prochlorococcus. Through the work of other laboratories (Rowher, pers. comm.), signature genes are beginning to emerge that allow for the phylogenetic analysis of phage types based upon the sequence analysis of one or a few conserved genes just as has been done for microbial biota using 16S ribosomal DNA. Building upon this work, we have the opportunity to compare the phylogeny of the host and prophage detected within different Prochlorococcus clones to determine the relative importance of vertical or horizontal transmission of phage within the Prochlorococcus community.
What are the relative abundances of Prochlorococcus prophage in natural communities?
Estimating the abundance of prophage in a natural community has traditionally been difficult due to the dependence upon culture-based techniques selecting at two levels (the culturability of the host and the culturability of the phage) and due to the unknown selection of an appropriate inducing agent to target "all prophage." Through statistical analysis of our BAC clone libraries, we will have the unique opportunity to be able to approximate the abundance of prophage within the Prochlorococcus community using culture-independent techniques.
Do prophage confer host cell fitness advantages and drive niche diversification of Prochlorococcus?
We know from other phage-host systems that prophage often encode virulence factors and / or novel genes that allow significant fitness advantages of a lysogenic (prophage-containing) cell over non-prophage containing cells. Detailed characterization of prophage from our BAC libraries will allow for the identification of genes encoding such factors that might drive the physiological diversification of Prochlorococcus ecotypes in oceanic systems.
Progress to Date
Diversity of 23S rDNA in the Plum Island Sound.
We have constructed and screened a large-clone library from a coastal environment by the methods outlined in the experimental approach. Using our recently developed protocol to reduce PCR-generated sequence artifacts (see above), we have found surprisingly low ribotype diversity in this environment. Although the screening is still in progress, we currently estimate about 277 ribotypes to be present in the library (Fig. 1). This allows us to put a first lower boundary on total gene content for this community. Genome size of free-living bacteria ranges from 0.98 to 9.4 Mbp. Taking E. coli as our model with 4.6 Mb and roughly 4,400 genes we can estimate a minimum total environmental genome of 277 x 4.6 = 1,274 Mb and 277 x 4,400 = 1,218,800 genes (some of so close as to be allelic, others distant homologs, or non-homologs). In comparison, the human genome, which has been sequenced, is 3,000 Mbp but is thought to have 30,000 or more genes. This suggests that environmental communities may be accessible by genomics. However, a critical but unexplored variable in this calculation is the degree of within-ribotype diversity of co-existing bacteria. It will be essential to estimate within-ribotype diversity to arrive at reasonable estimates of total diversity.
Capture of large genome fragments from the environment.
We have developed a protocol that will be used to capture large (>20 Kb) genome fragments from environmental DNA. The protocol was first optimized using Vibrio cholerae DNA that was completely digested with SmaI resulting in a fragment of 6.1 kbp containing the rRNA operon. Fragment capture with a specific, 23S rDNA targeted 70-mer oligonucleotide showed good recovery with 62 ng of specifically enriched DNA. This fragment was then cloned by the methods described below. Subsequently, we were able to recover similar amounts of a ~20 kb genomic fragment when V. cholerae DNA was spiked into DNA extracted from a natural community at 10, 1 and 0.1%. We anticipate being able to recover much larger genome fragments using partial digests of environmental DNA that has been size fractionated on pulsed field gels. As detailed below, we will ultimately use this method to obtain large fragments of DNA from uncultured organisms with unknown genome composition.
Proposed Approaches
Field Sampling
Chisholm already has an ongoing NSF project at the HOT and BATS stations (see Prior Support section), thus obtaining the samples from there will not be a problem. We also have an ongoing collaboration with Dr. Anton Post, a cyanobacterial expert at the Interuniversity Institute of Eilat, who has regular cruises on the Gulf of Aqaba (see letter of collaboration), thus facilitating our sampling there.
Sample Preparation
Cell collection and concentration. We will need a minimum of 2 x 109 Prochlorococcus cells for each BAC library (~ 20 liters of water); however, to ensure sufficient coverage, we will concentrate cells from 100 liters. Samples will be pre-filtered (1 m m pore size) to reduce concentration of larger, eukaryotic cells. The remaining cells will be concentrated by tangiental flow filtration and pelleted by centrifugation as described by Béjà et al. . The cell pellet will be frozen in liquid nitrogen.
Cell sorting. Prochlorococcus cells will be sorted from other phytoplankton and heterotrophic bacteria using the MIT flow cytometry facility, which is equipped with several MoFlo flow cytometers (Cytomation). As we have shown many times in our past work , Prochlorococcus has a unique flow cytometric signature that distinguishes it from other phytoplankton and heterotrophic bacteria, and we have sorted them from field populations for other molecular studies . The MoFlo instrument has high-speed sorting capability, and can sort up to 30,000 cells per second, which means we could get the requisite 109 cells in a 24-hour period.
If we stain the DNA of the community with a fluorescent stain like Hoechst, we will be able to cleanly sort the Prochlorococcus away from all of the heterotrophic bacteria. This would be the ideal approach, and we will use it if we can show that the stain will not interfere with the remainder of the analysis, or that we can remove the stain before the analysis without disrupting the DNA. If this approach fails, we can still greatly enrich the Prochlorococcus cells relative to the heterotrophs through sorting, and the "contaminating" heterotrophs should be easily identified in our libraries. Since statistically they will be the dominant heterotrophs in the sample, some exploration of their genomic identity could be quite interesting and we will treat this as an ancillary part of the work.
DNA extraction. Nucleic acids for diversity estimation by PCR amplification and cloning will be extracted using bead beating , which yields DNA from difficult to lyse cells including Bacillus spores. Although cultured Prochlorococcus cells easily lyse quantitatively, the bead-beating will serve as a reference for the more gentle nucleic acid extraction method used for BAC library construction. High molecular weight DNA for BAC construction will be extracted as described by Stein et al. . Cells will be embedded in agarose in syringes and lysed by extrusion of the mixture into lysozyme and detergent containing buffer. DNA will be retrieved by enzymatic digestion of the agarose and will be subjected to shearing (see below).
Diversity estimation
Outline. We will estimate the number of co-existing bacterial ribotypes, and, as a preparation for Prochlorococcus BAC construction, the number of Prochlorococcus genomes in our samples, by determination of the sequence diversity in several genes and genetic elements. This will allow us to decide the necessary number of clones needed in the Prochlorococcus BAC library for the desired 15 to 20 x coverage of co-existing genomes and will provide us with suitable molecular markers for identification/quantification of specific genotypes in environmental samples or culture collections. We will target genes that accumulate sequence change at different rate but are limited to genes for which good PCR primers are available. For the total community, 16S and 23S rRNA genes will be used, and for Prochloroccus the internal transcribed spacer (ITS), and the RNA polymerase and the recA genes will also be assayed. Diversity of each gene will be estimated from rarefaction of sequence diversity in PCR-generated clone libraries. We have previously done this for the bacterial community using the 23S rRNA genes (see above), and for Prochlorococcus using the ITS, which is single copy in Prochlorococcus, and have found 20 co-exisiting sequence variants . Since the ITS is considered hypervariable, we expect this approach to be possible for all genes.
PCR amplification and cloning
. All PCR amplification protocols will take into account recent insights into generation artifacts including formation of heteroduplex molecules, which we have recently found to be a potential major source of artificial sequence diversity . Thus, at least 10 replicates will be amplified for only 15 cycles to minimize skewing of the distribution of sequence types and accumulation of mutations and chimeric molecules. Reactions will be diluted 1:10 into fresh reagents and amplified for 3 cycles to remove heteroduplex molecules followed by pooling and cloning. We can measure the ratio of the different amplification products in the PCR and can extrapolate to the gene templates by estimating amplifications kinetics using our Constant Denaturant Capillary Electrophoresis (CDCE) apparatus. This provide important information for calculation of the necessary coverage of the different librariesPCR primers. For 16S rDNA, the standard Bacteria specific primers 27F and 1492R including recently published modifications. For 23S rDNA, our recently re-designed Bacteria-specific primers will be used. These are perfectly matched to all Bacteria 23S rDNA sequences in the Ribosomal Database Project (RDP) and amplified a set of 40 phylogenetically representative bacterial strains (Klepac and Polz, unpublished). For ITS, primers anchored in 16S and 23S rDNA will be used . For recA amplification, primers described by Eisen will be used. The gene for DNA-dependent RNA polymerase will be amplified as described by Palenik .
Diversity estimation by in situ amplification (polony formation). As a longer-term technology development project, we will adapt the new polymerase colony (polony) method of PCR amplification in thin polyacrylamide gels with one covalently immobilized primer (Mitra & Church, 1999*, see attached) for rapid diversity estimation of bacterial ribotypes. DNA extracted from environmental samples will be deposited at appropriate dilutions on glass microscope slides and amplified in situ. The resultant PCR colonies (polonies) will be hybridized or sequenced in situ for sequence identification (Mitra et al. 2002*, see attached). This would allow the simultaneous sequencing without prior cloning of thousands of polonies on the slides.
Library and polony screening and diversity estimation. All libraries will be screened by automated sequencing of clones with a single primer (RevPrep Orbit (GeneMachines) and 3700 sequencer). A complete sequence for several representative clones in sequence type will be obtained. In all cases, the success of the sampling process will be monitored by rarefaction analysis and the total number of sequence types in a sample will be determined by the Chao-1 estimator. Confidence intervals for the Chao-1 estimator will be calculated as described by Hughes et al. .
Phylogenetically ordered large genome fragment libraries.
Outline. We will capture large genome fragments from bacterial ribotypes to estimate within ribotype diversity and to assay genome structures of important uncultured members of oceanic communities (e.g., members of the "SAR" (Sargasso) cluster, which are dominant bacteria in all oceanic environments) or important sinks of carbon originating from Prochlorococcus (see below, functional diversity array). For this purpose, a 70-mer probe complementary to a highly variable region within the 23S rDNA of each selected ribotype will be constructed. For each ribotype, the captured DNA will be cloned and thus a set of phylogenetically ordered libraries generated. The inserts in each library will vary in size since the environmental DNA is incompletely digested and enriched for size above a 10 kbp cutoff. Furthermore, the library may contain a background of ribotypes that were captured non-specifically by the 70-mer probes. Thus, the initial characterization of the libraries will involve a four-step analysis protocol, which allows exclusion of non-desired clones. First, inserts will be sized by pulsed field gel electrophoresis. Second, inserts above 10 kbp will be screened by RFLP using hexameric restriction enzymes and ordered by similarity. The following groups of cloned inserts are expected: (1) same pattern, same size, (2) similar pattern, different size, and (3) different pattern regardless of size. Third, ribotype identity will be confirmed by sequencing of the 23S and 16S rDNA in the same set of clones and only identical ribotypes will be further analyzed. Fourth, the sequence in the flanking region (gene) downstream of the 23S rDNA will be determined in all clones containing identical 16S ribotypes. Two groups of clones are expected that contain (1) homologous flanking genes and (2) non-homologous genes. Our subsequent analysis will concentrate primarily on the first group since these clones stem identifiably from orthologous rRNA operons (see below). Preference will be given to clones with complete ribosomal operons (and complete operons will be sequenced).
Probe construction. Specific 70-mer oligonucleotides will be constructed based on alignments of 23S rDNA sequences recovered in our PCR-generated clone libraries using the GCG (Genetic Computer Group, www.accelrys.com) sequence editor. We have previously determined that tethered 70-mer oligonucleotides have very uniform dissociation behavior almost independent of the sequence (rRNAs have a limited range of GC-content) (Marcelino et al., unpublished). Thus, optimization of hybridization temperatures and conditions is not needed. For genome walking by capture, we will use PCR-amplified sequence stretches from the ends of the initially captured fragments. These should hybridize and capture homologous genes.
Capture of genomic fragments. Oligonucleotides are tethered to a linker oligonucleotide, which is biotinylated. Hybridization is carried out in solution and the hybridization product subsequently captured using streptavidin coated magnetic beads. The efficiency of this process is demonstrated in the preliminary results section.
Cloning. The captured single stranded fragments are cloned by attachment of linker oligonucleotides and subsequent partial second strand synthesis using Klenow. This enables either blunt end cloning or forced cloning via restriction sites introduced in the linker oligonucleotides. The plasmid containing the insert are then be transformed into E. coli host cells where the second strand will be fully synthesized. The efficiency of the process has been demonstrated (see preliminary results). To date, we have chosen the PBluescript II SK (+/-) Phagemid (Stratagene), which can carry up to 15 kbp, inserts but the process can be adapted for other plasmids, including BACs.
Sequencing and analysis. We will aim for the initial sampling of captured genomic fragments of 10 unique ribotypes. Preference will be given to captured fragments that contain near complete rRNA operons. Within ribotype diversity and diversity among closely related ribotypes (<5% 16S rRNA sequence divergence) will be examined by sequence comparison of genome regions flanking strictly homologous (orthologous) rRNA operons. These will be identified by the presence of at least one homologous flanking gene. By primer walking, we will sequence the flanking regions of a set of representative fragments. Contigs will be assembled and sequences will be edited and aligned using Sequencher (Gene Codes, Ann Arbor, MI) and GCG v.10 (Acelrys), and open reading frames will be identified. BLAST similarity searches will be conducted to identify and characterize homologs. Sequence divergence in flanking genes will be estimated for synonymous (KS) and nonsynonymous (KA) sites using DIVERGE (GCG) as estimators of selection. Furthermore, we will look for evidence of recombination, gene loss or rearrangement (as described below).
Functional genotype multiplexing
We will develop functional genotype multiplexing, a new technique based on in situ amplification methods (Mitra and Church, 1999*, Mitra et al. 2002* see attached), that allows the rapid simultaneous phylogenetic identification of organisms and determination of presence of specific functional genes. This will be done by embedding in thin acrylamide gels bacterial cells that have been made permeable by enzymatic digestion and short fixation, and by subsequent in situ amplification in the gels. Embedding in the gels allows high multiplicities of cells and probes to be inspected without formation of crossover products or heteroduplexes during the PCR since each cell's amplification is compartmentalized away from other cells. One previous source of variability is PCR without prelysis. Some environmental bacteria are hard to lyse, enzymatic digestion is used to make them permeable in the context of the immobilization polymer the cell DNAs remain separate. The polony format seems to compensate for differences in amplification in another way, which is that the amplicons, which start early or go faster switch from exponential to slower growth (cube-law) while the slower ones stay exponential. This is expected due to saturation of the core of the polony. Several primer pairs can be included in the amplification mixture embedded in the gel, some of which can be ribotype or phylogenetic group specific. Alternatively, rRNAs can be amplified with universal primers and different bacterial phylogenetic groups identified by hybridization. Identification of colocalized genes is by labeling of primers with different fluors and subsequent detection of specific color mixtures. Our current experiments on FACS sorted mammalian T-cells are expected to be considerably more challenging than the microbial cells and will allow us to work out methods to get acceptably low "false-negative" rates due to limitations on single molecule PCR, which we have already pushed to over 80% efficiency per molecule.
Prochlorococcus
BAC LibrariesOutline. BAC libraries will be constructed for the samples described in the section above and the size of the BAC library will be determined by the diversity estimates. We currently estimate based on ITS diversity that ~20 ecotypes co-exist. This would entail a library size of 10,000 clones assuming 20 x coverage. At least one of the BAC libraries will be fingerprinted to assemble large contiguous genome fragments of the co-existing Prochlorococcus (see sequence analysis). However, we will strive to include a second library if the encountered diversity and costs allow.
Vector and host: We will use HS996, which is based on the most commonly employed host DH10B for BAC libraries, carrying an additional phage T1 resistant mutation. The resistant mutation prevents the destruction of the libraries due to host lysis by possible phage T1 contamination in laboratories. We will employ pIndigoBAC536 as a cloning vector. It carries four unique restriction sites at the cloning region; HindIII, BamHI, EcoRI and Eco72I. The Eco72I site will be used for blunt end ligation of sheared DNA sources.
Preparation of sheared DNA: BAC (fosmid) cloning from sheared DNA requires careful preparation of sheared DNA with properly terminated ends: the blunt ends must have 3’-OH and 5’ P terminated ends. Over the last two years, we have established a highly efficient and reproducible method for obtaining sheared DNA fragments of 30 to 100 kbp. The agarose DNA plug (100 to 200 ul in volume) is melted in an Eppendorf centrifuge tube by heating at 65 C for 15 min, and digested by agarase to extract DNA. The DNA is sheared by vortexing at maximum speed for 60 sec and by repeated passing through a 26 gauge needle. The resultant sheared DNA accumulates sharply at apparent size of 50 kbp. Although the structure of the ends of these sheared DNA are unpredictable, we found that a simple fill-in reaction by T4 DNA polymerase and phosphorylation by T4 ligase create ends in proper configuration for blunt end ligation.
Library construction:
We will make four libraries with a currently estimated size of 20,000 clones (average insert size 36 kb). This estimate is based on a genome size of Prochlorococcus of 2 Mbp and 20 ecotypes in the flow sorted fractions to yield 15 to 20 X coverage for a complete physical map of each genotype. However, the exact coverage will take into account the estimation of ratios of gene targets as a proxy for distribution of the different genomes obtained by CDCE-PCR analysis in the diversity. screening. We will make two copies from each library; one working copy and the other for replication. The libraries will be stored in 384-well microtiter plates at –80 C. We have developed a series of quality control procedures to maximize library construction and to ensure the size of the inserts is in the required range (30 kb to 55 kb). For example, we will check the percentage of the empty (no insert) clones and the size of the inserts in the libraries. (We found that virtually all of the colonies generated by lambda packaging have inserts.) Twenty colonies generated from each ligation mixture will be checked for the apparent size of the inserts by pulsed field electrophoresis.Making filters for colony hybridization: We will make initially 10 sets of filters for colony hybridization (to be distributed among the collaborators). 40,000 colonies (20,000 unique colonies x 2) can be printed on one 20 cm by 20 cm nylon membrane by Genetix Q-bot. Each set consists of four such sheets. After processing and fixing DNA on the membrane, they will be shipped to the investigators. More will be made as needed.
Fluorescent Fingerprinting of BACs
NOTE: The fingerprinting section describes work proposed in the collaborative NSF Biocomplexity proposal by Chisholm, Polz, Shizuya and Olsen. It is repeated here for clarification but is not part of the budget for this proposal. The fingerprinting method that Shizuya and collaborators have developed is based on end filling with fluorescently labeled nucleotides as described in detail in . We use a class IIS restriction enzyme, HgaI, which cuts DNA five bases away from the recognition site and generate a 5’ overhang having five unknown bases. These unknown bases can be sequenced with the recessed strand serving as primer and using modified fluorescent dideoxy terminator sequencing reagent (now available from Applied Biosystems). In order to accurately determine the size of each fragment, the fifth dye is included in each lane. With this in-lane standard, each fragment is characterized by both the size and the sequence of its terminal five bases. (To generate fragments small enough to size on automated sequencers, a four-base blunt cutter, RsaI, is included in the protocol. This method greatly increases the power to detect minimum overlap between clones (10 - 15 % overlaps as opposed to 50 % in agarose-based fingerprinting), and to assemble highly accurate contigs.
We use the modified FPC program to cluster clones into overlapping groups. The program was originally developed by the Sanger Centre, and based on an algorithm calculating pairwise probability of coincidence, which specifies a random chance of non-overlapping for two clones that share a certain number of fingerprinted fragments. However, since it uses the information solely based on the size, it is not usable for five colored fingerprinting fragments. To be able to use the color information (base sequence information), we have modified the program. The modified program is now available from the site: www.discoverybio.com.
We have successfully tested the method on 555 BAC clones from human chromosome 16p13.1 to 16p11.2 that could previously not be assembled using marker-based hybridization and agarose-based fingerprinting due to regional duplications. Because we expect high homology among DNA from Prochlorococcus species in the libraries, five base terminal sequence information together with the size information will greatly enhance the accuracy of the contig assemblies and generate highly accurate physical maps.
We estimate that it will take three months to prepare BAC DNA from 20,000 clones, and require an additional six to nine month to fingerprint by a 3700 and/or 373 ABI DNA sequencer. Data assembly will take three months to complete.
Comparative genomics of Prochlorococcus BAC libraries
Outline. Four BAC libraries will be constructed, one of which one will serve as the ‘reference’ library. The reference library will be completely fingerprinted (depending on matching funding from NSF, see above) or sequenced with the goal of assembling complete genomes. The success of assembly will depend on how many genomes co-exist and how similar these are to one another. If the genome diversity is moderate we will be able to tile overlapping BACs and obtain a detailed picture of overall genome architecture and superimposed variation. If genome diversity is too high to assemble contiguous BACs we will establish anchor points in the BACs by probing with highly conserved genes, primarily from informational genes since these have lowest probability of lateral gene transfer . In this case, the portrait of genome evolution will be of variations in local genome structure (at a minimum the size of BAC inserts) but will nonetheless provide an unprecedented set of data for interpretation of mechanisms of diversification and selection. The other BAC libraries will be a rich source of comparative data with the background of relevant environmental variation and substructure of the metapopulation. The presence/absence and sequence diversity of specific genes and pathways will be compared among all BAC libraries (Table 3). This includes genes and pathways that have already been shown to display key differences in environmental adaptations, including photosynthesis, inorganic carbon fixation, nutrient uptake (N and P), temperature regulation, and organic carbon metabolism. However, an important asset is the current development of DNA microarrays that will be used for interrogation of gene expression patterns under varying environmental regimes. Specific genes will be identified in the BAC libraries by hybridization of the filters made from BAC colonies. Since multiple probes can be used per hybridization and the filters can be reprobed at least 10 times we could search for at least of 500 genes. More filters will be printed if needed.
Target genes for comparative analysis. As outlined above, several genes and the pathways that are associated will be targeted either for anchoring the BAC sequencing or for comparison of the BAC libraries from the different environments. Anchoring genes are those that have low probability of lateral transfer due to the high numbers of interactions of their gene products in the cell and those that encode important pathways (e.g., photosynthesis and nutrient uptake). Genes for comparison of the BAC libraries are those that allow testing of the questions of diversification of the populations and the influence of different selection regimes. Thus, besides information about ecological function, we will include genes with differences in expression level for interpretation of codon bias, and genes under strong or weak environmental selection. A list of target genes (Table 3) has been drawn up from currently available information based on genome comparison of the Prochlorococcus ecotypes MED4 and MIT9313; however, continuing genomic comparison and information based on environmental regulation of sets of genes as determined by DNA microarrays will serve to supplement this list.
Gene Diversity and Divergence. Analyses of informational and central metabolic genes will provide a baseline of sequence diversity and relative divergences at the DNA versus protein level. Establishing such a baseline within the biological system is very important, since the broad range of G+C contents of MED4 and MIT9313 within this relatively tight phylogenetic group indicates one or more shifts in "G+C pressure" (e.g., . Thus, it will be important to characterize (in essence, calibrate) these measures within the low-G+C group, within the high-G+C group, and between the groups. With this background it will then be more meaningful to interpret the diversity and DNA versus amino acid divergences of other proteins, including those
Table 3. Candidate genes for the comparative analyses. The number in parenthesis after the gene name indicates relative degree of expression in MED4 when grown under continuous light (Zinser, unpubl.). E=Environmental, H= Housekeeping. Yes and No indicate presence or absence in the genomes.