Excerpted from CEGS Grant Proposal Jun 2002
Aim 2:
Mitra & Church (overall PI & Director: Wu)
C/D2. In situ measurements of polymer modifications.
We propose to extend in situ RNA/DNA analyses to measure
changes at the level of DNA (mutation (2a), recombination(2b), and methylation
(2c)), RNA (alternative ends (2d) and splicing (2e)), and protein epitope
combinations (2f). For each we will use
polony concepts and aim for subcellular and single-residue (modified nucleotide
or amino-acid) resolution. Another
unifying theme is the assessment of for "cis-ness". For example, which alleles are in cis in DNA
haplotypes, which exons are in cis in a spliced RNA molecule, which protein
modifications are in cis in a protein molecule? Here the use of single
molecules is crucial for interpretation, as a population average could be very
misleading. Analogously which of the
above molecular features occur in the same cell (in contrast to a cell
population average)?
The key
problem in genomics is that sequence comparisons (and SNPs) alone do not
predict functioning of physiological systems accurately. Functional genomics
should do better, but is weak and costly due to the enormous multiplicity of
cell-types, gene products, and interactions and cross-reactions among network
components. This is a very general
problem since nearly all organisms form complex cellular patterns from
microbial communities to developing plants and animals. How can we
cost-effectively monitor the comprehensive set of cell types? Functional
genomics of humans is further complicated by difficulty in accessing key cell
types (e.g. brain tissue). Single
nucleotide polymorphisms (SNPs) do not necessarily predict diseases with
acceptable precision for individual patients even if the statistics are
convincing in populations. Instead of "genetic
linkage/association" one would favor "functional association".
A desirable
long-term scenario for clinical diagnostics illustrates key research goals:
Obtain cells from a patient (Aim 3). Use
automated microfluidic differentiation system (mDS, Aim 4) to create and
display a variety of cell-types in a compact format. Stain with multiple
oligonucleotides and/or antibodies, multiplexing probings and fluorophores to
obtain allele-specific and cell-type-specific phenotypes. Use this data to computationally prioritize
significant deviations of the patient relative to normal variations in the
population. As can be seen from Aims 3 & 5, common dosage effects have
significant phenotypic consequences. Linear 1.5 fold increases or 2-fold
decreases must be discriminated from feedback pushing closer to 1-fold, or
amplification beyond 1.5 or 2.
Non-causative SNPs can be used to quantitate allele-specific effects
(relevant to Aim 1). This allows for
detection of small expression effects in heterozygotes (a 10-fold
allele-specific decrease corresponds to only a 1.8-fold decrease in total RNA
for that gene).
Preliminary results Single-molecule cis-typing: We are
developing new technologies which exploit intrinsic advantages of single
nucleic acid molecules for DNA haplotyping and RNA exon-typing. These use the polymerase-colony, a.k.a.
"polony", hybridization and allele-specific fluorescent-base addition
described in Aim 2d. This can be
generalized to include multi-base extension (a.k.a. "in situ sequencing").
An example of such sequencing is given in the figure below. Similarly, multiple rounds of hybridization
have been used to determine the exon composition of single RNA molecules (and/or
single cells) in Aim 2e, below.
We also have
developed a variety of image/data processing and network modeling algorithms
(see Aim 5 preliminary results). The
problem of computational image alignment and cost will be greatly reduced by
increasing the multiplicity of probes per embryo mount. Image processing demands
are reduced in various ways e.g. by fluorescent color ratio assays and internal
controls for alignment.
2a. DNA haplotyping will be developed to
determine which combinations of DNA polymorphisms in cis are associated with
inherited allele-specific expression seen in Aim 1. For example, in collaboration with Brad Hyman
(MGH Neurology) we are assessing if the reason for ApoE3/E4 heterozygous
variation in Alzheimer's phenotype is related to level of expression of the two
protein forms. Since there is significant
tissue specificity, a surrogate tissue approach is unlikely to work and even
tissue-specific and allele specific protein abundance correlations with disease
could be the result of a protein difference rather than caused by a
transcriptional difference. Hence the need to haplotype over
the distance between the protein change and the promoter/enhancer
polymorphisms.
2b. Recombination and combinatorial tags: Three
types of recombination are of potential interest. One is VDJ recombination which determines a
large fraction of the epigenetic state of B and T cells as well as the
specificity of the antibodies in Aim 2f.
A second is class switch recombination (CSR) which could act as a
lineage reporter (in addition to methylation below). The third system is homologous recombination
(meiotic, mitotic or transfection) as mentioned in Aim 2a, 2f and 5. By way of preliminary results, our lab
invented the use of libraries of short quasi-random recombinants as unique tags
[1]. These recombinants are important as
a means for uniquely tagging progenitor cells for developmental lineage studies We were
involved in the first such lineage study which was developed for retinal and
cerebral cortex [2]. Another example of
recombinant libraries with quasi-random short tags would be the antibody
libraries in Aim 2f. Here the sequence
of the randomer not only is a tag but also a web-shareable definition of the
“reagent” analogous to the SequenceTagged Sites (STS) and Expressed Sequence
Tags found useful in the Genome Project (HGP).
The switch of the immunoglobulin isotype from IgM to IgG, IgE or
IgA is mediated by class switch recombination (CSR). Activation Induced Cytidine Deaminase
(AID) is indispensable in both CSR and somatic hypermutation. Ectopic expression of AID induces CSR in an
artificial switch construct in fibroblasts at a level comparable to that in
stimulated B cells. The features of junction points were similar to those
observed in physiological CSR: no consensus sequences around the break points, no
long homology between two S sequences at the junctions, and frequent mutations
in the proximity of the junctions and dependent on transcription of the target
S region, as in endogenous CSR in B cells [3].
Thus
the CSR-based recombinations seem to result in unique sequences. We propose that combining expressed switch
regions with a transgenic AID gene under control of a chemically-inducible
(e.g. anhydrotetracycline) promoter would allow us to pulse the tagging of all
cells in a developing mouse. Using an adjacent constant primer for polony
sequencing of a sufficient number of variable bases after recombination would
be sufficient to constitute a likely unique tag (by virtue of it including both
a recombination junction and mutations).
2c. DNA methylation and lineage
can be
assessed by bisulfite deamination of cytidines and sequencing. Methylation allows (partial) lineage
reconstruction of the history of cell-divisions, migrations, and epigenetic
changes for each cell as embodied in recent algorithms [4,5].
The primers required for bisulfite analyses have additional constraints beyond
those that apply to genomics more generally [6,7].
These are embodied in a public primer resource [8].
We will use in situ polonies amplified on embryos or
cells as in Aim 1 or mDS in Aim 4. This will allow
retention of the in situ geometry and
lower costs as in aim 1c, 2b,d-f. Yet another
advantage of caging cells and nucleic nucleic acids
used in polony amplification is reduction of loss during extensive washing [9]
allowing "amplified sequences of a single copy gene from as little as 50
pg of bisulphite treated chromosomal DNA (~10 individual cells)." We
expect to do even better because our reaction volume is a million times
smaller, our PCR products ten times shorter, and we might go to milder
treatment than 4 h at 50C pH5 due to the immobilization effect, e.g. recent
results claim that "0C for 1 h resulted in 98% conversion" to dU [10].
2d. In situ RNA sequencing and RNA 3' polyA end choice. We will combine the in situ methods used in Aim 1a,b with allele specific methods of 1c.
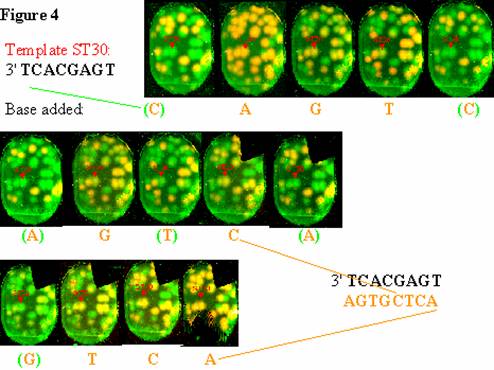 This
will allow us to measure two DNA or RNA alleles close in space (as in
transvection) as well as quantitative differences in expression of the two
alleles as can occur in regulatory mutations and/or graded versions of
epigenetic phenomena. Aim 1c describes a
way to do this on the diluted contents of single cells. Aim 2d attempts to do the same in situ in the case where a polymorphism
lies near a polyA end. It also allows
discovery and quantitation of
RNA 3' end choices. This
is somewhat analogous to Long-SAGE/MPSS [11,12] but
with the huge advantage of being in situ
capable.
This
will allow us to measure two DNA or RNA alleles close in space (as in
transvection) as well as quantitative differences in expression of the two
alleles as can occur in regulatory mutations and/or graded versions of
epigenetic phenomena. Aim 1c describes a
way to do this on the diluted contents of single cells. Aim 2d attempts to do the same in situ in the case where a polymorphism
lies near a polyA end. It also allows
discovery and quantitation of
RNA 3' end choices. This
is somewhat analogous to Long-SAGE/MPSS [11,12] but
with the huge advantage of being in situ
capable.
The
signal-to-noise for typical FISH is not currently compatible with Single
Nucleotide Polymorphism Extension (SNuPE) assays [13]. We have developed methods for in situ amplification of the number of
template molecules (centered on one initial molecule) to enough copies to get
robust allele determination [14]. We
call this general class of amplification products polonies (short for
polymerase colonies, see also Aim 1c).
Although we and others have used rolling circle amplification [15] or in situ PCR [16-18], these do not
cleanly lead to a single primed template as needed for single-base or
multi-base extension assays. Although single-molecule extensions can be
visualized [19], the added robustness from dual primer constraints in polonies
is significant. Instead, our polony method begins with embedding the target RNA
or DNA molecules in polyacrylamide such that one PCR primer is covalently part
of the polyacrylamide gel via an acrydite moiety [14], while the other primer is free to
diffuse. This allows removal of one
strand for subsequent hybridization and/or polymerase extension. In this method, polony size is reduced by
increasing acrylamide concentration, template length, decreasing number of
cycles. The lower limit so far has been
10 micron diameter polonies. An
alternative would be to immobilize both primers. Three small barriers prevented this
originally, but probably no longer.
First, the small amounts of free primers must be as low as
possible. We now accomplish this by
pre-electrophoresis. Second, the presence of the second strand would be a
potent (intramolecular) competitor. To
solve the latter problem, we will test photocleavable (PC) linkage of one
primer to the matrix by incorporating acrydite (Apogent) and PC-spacer
phosphor-amidites (Ambergen)
[20,21] on the 5' end of the oligo in the penultimate
step of oligonucleotide syntheses. We have already demonstrated the use of PC
linkages between dNTPs and their fluorophore (using similar PC nitrobenzyl groups from Ambergen). The third problem is the fact that the rate
of spread of the polony with double immobilization is limited by template
length. We will systematically test lower acrylamide and/or bis-acrylamide
concentrations to allow the effective radius of to be larger.
Automated multiplex in situ sequencing and microscopy. As an extension of the in situ methods (in 1a, 1b, 2b and 2c), we will push the limits of multiplexing , resolution and data integration. This will mainly be done in the context of
sequencing 3' ends of mRNAs in situ,
but we will also see to what extent the same concepts can be applied to
methyl-CGs (Aim 2c), RNA allele typing (Aim 1c), and exon typing (Aim
2e-f). In addition to the limits of
resolution due to the polonies technology itself, there are limits set by
optics and signal-to-noise and scan speed. Very fast scanning is available for
microarrays at 3 to 5 micron pixel size.
However, specialized resolutions in the sub-100 nm range have also been
demonstrated [22,23]. Multiphoton fluorescence excitation provides
another dimension [24-26]. Some of this will require developing quantitative
microscopy with full slide scan and autofocusing very analogous to the array
scanners, but more challenging due to resolution required. Computational
integration of adjacent sections is also important. We anticipate this type of alignment to be
easier to automate than our "one- or two- color" alignments because
of the rich RNA profile available for each voxel in
the images. "Optical projection tomography" has been used as a tool
for 3D microscopy and gene expression studies [27].
2e. RNA exon-typing is a key example of how the
cell exploits combinatorial opportunities.
The fraction of genes showing alternative RNA splicing has risen from 5%
to 50% since 1994 [28]. Many of these alternatives have huge effects on the
physiology and pharmacology of the resulting proteins. Aim 2a.will allow us
to accurately determine alternative spliceform distributions without
full-length cDNA clone sequencing. This will be done by amplifying potentially
variable exons and then sequentially or simultaneously probing for each
putative exon. Exons can also be
discovered and quantitated in situ by
polony sequencing from a known exon into whatever is
3' or 5' of it (see Aim 2d). Aim 2a will
also feed into the Aim 5a goal of aligning in
situ images exploiting rich profile data.

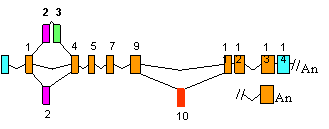
Figure 2e. Alternative splicing in
Tau.
By exon-specific hybridization to polonies amplified from single RNA molecules
using exon 1 & 11 primers. Mutations
in the human tau gene cause frontotemporal dementia and Parkinsonism associated
with chromosome 17 (FTDP-17). One of the major disease mechanisms in FTDP-17 is
the increased inclusion of tau exon 10 during pre-mRNA splicing [29].
2f. Protein multiplexing in situ. Clearly
"affinity proteomics" is a huge gap in the current application of
genomics to biology. Antibodies are
crucial for in situ quantitation,
antibody-arrays,
immunoselection/precipitation (IP), etc. The importance is that
an enormous fraction of the cell-type specification and disease is specified by
differences at the post-translational level.
How can we even begin to get the reagents since they are not currently
considered as "designable" as reagents for RNAs? We will develop methods for obtaining protein
epitopes simultaneously with cognate oligonucleotide-tagged-antibodies. We will strive to make them suitable for
highly-multiplexed, single-molecule polony-tag readouts and discovery of novel
epitope combinations. We will make versions which are "humanized" to
be compatible with human immune systems, or easily reversible as needed in Aim
4d. Finally they will be shareable electronically as described in aim 2b.
Preliminary results. We have three relevant
activities. First, we have been among
the first to establish large-scale proteomics [30] as well as algorithm
development and crosslinking experiments [31].
We believe that we have the first proteome of an organism
"completed" in the sense of finding peptides for every reasonable
open reading frame. Second, we work with the Harvard Institute of Proteomics
and FlexGene Consortium [32] with a goal of producing
at least one full-length expression cassette for each annotated human
gene. The cassettes are designed to be
easily/automatically moved into dozens of vivo or in vitro expression system. We have adapt
lambda-red [33], yeast homologous recombination [34] and Creator in vitro
recombination [35] for this purpose. In collaboration with Rick Boyce (MGH
Neurology) we
are exploring a particularly safe and efficient VSV-Baculoviral expression
system for these cassettes in embryonic stem cells and neuronal derivatives
[36] .
Third, we (
Aim 2f. Experimental.
Combinatorial
chemistry, amplification, structure design and arrays are well-established and
useful for nucleic acids but other classes of ligands are more useful for
protein affinity measures. Hybrid
molecules capture the advantages of each.
The idea is that by attaching nucleic acid tags to reagents which bind
proteins one can amplify, probe and/or sequence the tags in situ to quantitate a multiplicity of proteins in an array or
tissue. The attachment can be to
affinity agents (e.g. antibodies, small ligands, lectins), which would bind to
cell arrays or tissue sections or protein arrays and after washing be detected
by nucleic acid methods (RCA, PCR, FISH).
Simple defined-length linkers can be used to turn moderate affinity agents
into an assembly, which has higher avidity and/or specificity. These assemblies can combinatorially explored
(created and screened) by annealing libraries of oligonucleotide-conjugated
ligands (e.g. active site agents, peptide loops) followed by polony analysis of
the tag composition (and possibly order).
One set of
methods that we pursue accommodates natural cDNAs without individual
manipulation of the stop-codon region of the cDNA. In addition to the demonstrations above using
(i) polyprotein and (ii) purified translation lacking release factors,
additional approaches include (iii) inhibitors of translational elongation or
release, (iv) dominant negative inhibitory mutant release factors, (v)
antibodies to release factors for inhibition or immuno-absorption subtraction,
(vi) suppressor tRNAs added to the vitro reaction to allow extension the last
20 amino acids out of the ribosomal exit tunnel, and (vii) finally, the nascent
chain could be ensnared by a binding motif on one of the ribosomal proteins,
e.g. a biotinylation motif peptide 14-mer BirA substrate[41] fused to the ribosomal-protein L22
N-terminus, which is free and near the exit channel of the nascent peptide
chain [42].
Peptide pair selection 1. Such expression pairs would
be useful for stimulating and measuring cell surface receptors (as in Aim 4), as well as
proteomics generally. We will
gene-fuse Next
a library of single-chain antibodies randomized in the appropriate variable
residues regions will be made from a prototype vector using partially
degenerate oligonucleotides. "Camelized human single domain antibodies" [43]. 5'-GCCCCAGATATC AAA (MNN)9GCA(MNN)10 TGC TGC ACA GTA ATA-3' (19 randomized codons
in bold) will be our initial target, as they are preferred as sterically small
(14 kdal) antibody probes and extraordinarily compatible with enzyme active
sites [44]. Human framework antibodies have the advantage of compatibility with
human immune systems for potential downstream diagnostic and therapeutics
applications. Efficient tumor targeting has been observed using single domain
VHs with dissociation constants of 2 nM to 65nM [45]. The camel Ab domains
have also proven useful in transmigrating across human blood-brain barrier
endothelium [46].
 Libraries (non-camel scFv) made entirely in vitro without animal or bacterial
cells with an initial complexity of about 2e9 have been screened using
mutagenic PCR ribosomal display to obtain antibodies "with monomeric
dissociation constants as low as 82 pM" [47]. N-terminal gene-fusions of the antibody with
a monomeric avidin [48] as the bridge will allow the antibodies to bind and be
released from the ribosome. The ribosome
display library will finally have a library of protein coding fragments. These fragments could be random exons,
designed codon peptides (see below), or membrane-bound polysomal cDNAs from
embryonic stem cells and differentiated derivatives. Various points within the Ig constant
scaffold can be replaced with very specific protease sites, e.g. factor Xa
site, IEGR, cleavable by taipan snake venom [49]. This allows release of the ribosome display
from the selection-solid-phase and also helps produce the reversible
visualization antibodies needed in Aim 4.
Libraries (non-camel scFv) made entirely in vitro without animal or bacterial
cells with an initial complexity of about 2e9 have been screened using
mutagenic PCR ribosomal display to obtain antibodies "with monomeric
dissociation constants as low as 82 pM" [47]. N-terminal gene-fusions of the antibody with
a monomeric avidin [48] as the bridge will allow the antibodies to bind and be
released from the ribosome. The ribosome
display library will finally have a library of protein coding fragments. These fragments could be random exons,
designed codon peptides (see below), or membrane-bound polysomal cDNAs from
embryonic stem cells and differentiated derivatives. Various points within the Ig constant
scaffold can be replaced with very specific protease sites, e.g. factor Xa
site, IEGR, cleavable by taipan snake venom [49]. This allows release of the ribosome display
from the selection-solid-phase and also helps produce the reversible
visualization antibodies needed in Aim 4.
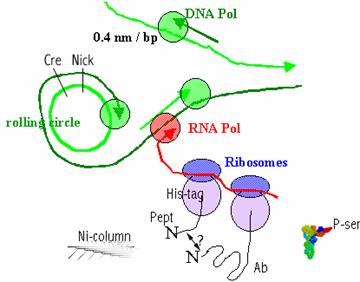
Figure 2f. (right)
Only the ribosome-display vectors in which the released peptide binds to the
antibody will bind to the Ni-column. A
positive round with a P-ser-tRNA (see modified peptides section below) is
shown, followed by protease release and a counter-selection round with a
ser-tRNA replacement and then a second counter-selection round with P-ser but
replacing other aminoacids that to create a peptide allele, splice-form, or
gene-family relative. These three rounds
could be done on a large library of peptide-antibody bicistrons simultaneously. To obtain high affinity antibodies,
previously successful whole-gene mutagenesis [47] could be enhanced by focusing
on the desired variable regions by use of specifically primed error-prone
polymerases (we have been studying these in collaboration with
Peptide pair selection 2. The above selection requires
that the first peptide of a pair in a bicistronic mRNA must bind to its
mate. A complementary concept,
is that two peptides (or antibody) bind to each other or to a third surface and
the proximity is "recorded" as a hybrid nucleic acid. There is some
precedent for this is methods based on ligation of aptamers [51] or rolling
circles [52]. The idea is that is two affinity tags need
to be near enough to one another to prime polymerase or allow a ligation to
occur. This is the protein equivalent
of Aim 2d (DNA proximity assay). There
are also conceptual similarities to protein FRET [53],
however our proposed method should have higher multiplex capability and
robustness. This method has the
advantage over single tags that it is resistant to non-specific binding because
it requires two tags to get a signal. The affinity between the two nucleic acid
tags is tuned so that they are weak enough stay apart in solution but strong
enough to bind together when kept in close proximity in the presence of
polymerase or ligase , e.g. 10-mers [51]. One way to accomplish this that ties in well
with the overall thrust of Aim 2f is using polyribosomes (from rolling circles
optionally) lacking release factors (as above) having free mRNA 3’ ends with
tags which co-prime RNA-dependent RNA polymerase followed by RT-PCR. Another is to set-up recombination between
the two DNA circles. This could result
not only in a signal of the cis-ness but also an immediately useful construct.
Epitope- or allele- specific mRNAs for antibody
selection: We are collaborating under separate funding
with a group at Univ. Houston (Linxiao Gao) and one at
Modified peptides: Alternative genetic codes [39] are more
straightforward to program with pure in vitro translation systems making short
designed peptides than for large genomes and more natural sets of translation
factors because one has complete control over codon usage and tRNAs
present. The new tRNAs
can be charged chemically [57] or by altered aa-tRNA-synthetases [58]. Examples of potentially useful tRNAs would be phospho-Ser/Thr/Tyr, methyl-Lys, methyl-Arg,
and acetyl-Lys or enzymatically resistant versions of these. For example, in
the histone "code" [59], 11 of the N-terminal 18 amino acids of
H3 are modified in many combinations of
the above aminoacids (see section B. c.).
For in situ analyses, peptides
displaying systematic modification combinations or antibodies directed at them
should be "programmable" using the above in vitro system. In a challenging project and complementary
source of selection, we will bind the antibody library to the differentiated stem cell arrays
from Aim 4 and release with the specific protease as above. We will try
polyprotein antibody libraries, a no-termination-codon library, and/or the
no-ribosome (puromycin) approaches.
Phosphothioate rNTPs will be used if RNAse resistance is desirable. The
intention is to broaden the scope to include post-synthetic modifications such
as glycosylation.
Specificity. Reagents which are specific to one combination of
post-synthetic modifications, or one isoform or allele
type are a holy grail with multiple uses in analysis and modulation of
activity. For each of the above antibody
selection methods, selection and screens for antibody specificity is important.
One option for would be to turn the selected library into a set of polonies and
screen for those which bind only their own peptide as assayed by FISSEQ. A
second option is to run the selection twice with different genetic codes
(above). A third is using antibody
combinations (as described above). A
fourth option is only available in conjunction with the synthetic 75-mer
programming is choosing peptides most likely to be unique in the first
place. It should be noted that when
using non-standard genetic codes both the peptide and the antibody will use the
altered code unless design or selection prevents that.
Proteomic cis-allele or cis-modification measures: Measures in a heterozygote are more reliable
if done in an allele-specifically than in the usual pooled measure because the
signal is undiluted (for example, a 2-fold change in the internally controlled
ratio is going to be better than the externally compared ratio of 1.5 fold in
comparing the rare heterozygote with the common homozygote. Doing this at the protein level is more
accurate than at the DNA-SNP level since many currently unpredictable steps lie
between the causative SNPs and the final protein levels. In the embodiment
where one tag is for an active-site-directed agent and another tag is directed
against a common protein surface polymorph-ism, one can assess allele specific
differences in protein function. This is analogous to Aim 2b, which assesses
allele-specific differences in RNA levels. The extension of HEP to protein
function is a step forward in accuracy of assessment of the function of new
SNPs by leveraging old SNPs into functional assays.
C/D4. Voldman, Badarinarayana, & Church.
a) Demonstrated multiple sequential
affinity labeling in flow
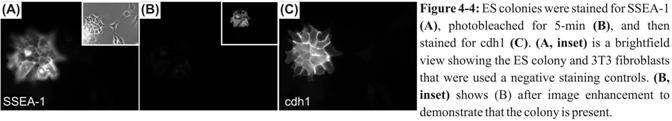 To automatically and viably assess
the phenotype of the differentiating cells in our mDS, we will use affinity agents to sequentially probe for
various cell-surface markers. We have demonstrated
a first step towards this goal by sequential labeling ES cells with two
antibodies in a flow system, photobleaching in
between the two (Figure 4‑4). Furthermore, we used indirect immunofluorescence with the same fluorophore, demonstrating that our
ability to multiplex in time obviates the need to multiplex colors and thus
dramatically extending the number of targets that we can probe. We will
adapt this scheme to the mDS, allowing us to assay surface marker
expression in living cells (Aim 4c).
To automatically and viably assess
the phenotype of the differentiating cells in our mDS, we will use affinity agents to sequentially probe for
various cell-surface markers. We have demonstrated
a first step towards this goal by sequential labeling ES cells with two
antibodies in a flow system, photobleaching in
between the two (Figure 4‑4). Furthermore, we used indirect immunofluorescence with the same fluorophore, demonstrating that our
ability to multiplex in time obviates the need to multiplex colors and thus
dramatically extending the number of targets that we can probe. We will
adapt this scheme to the mDS, allowing us to assay surface marker
expression in living cells (Aim 4c).
D
References for Specific Aim 2
2-1 Church,
G.M., and Kieffer-Higgins, S. 1988. Multiplex DNA sequencing. Science 240:
185-188.
2-2 Walsh,
C., Ryder, L., Cepko, C., Church, G.M., and Tabin, C. 1992. The
dispersion of neuronal clones across the cerebral cortex. Science 258:
317-320.
2-3
2-4 Ro, S and Rannala,
B. 2001. Methylation patterns and mathematical models reveal dynamics of stem
cell turnover in the human colon. Proc Natl Acad Sci
2-5 Yatabe Y, Tavare
S, and Shibata D. 2001. Investigating stem cells in
human colon by using methylation patterns. Proc Natl Acad Sci USA
98:10839-44.
2-6 Selinger,
D., Cheung, K., Mei, R., Johanson, E.M.,
2-7 Wright,
M and Church,G.M. 2002. An Open-source Oligonucleotide Microarray Probe Standard for Human
and Mouse. Submitted to Nature Biotechnology.
2-8 Li, L.-C.
2002. MethPrimer
- Design Primers for Methylation PCRs http://itsa.ucsf.edu/~urolab/methprimer
2-9 Olek A, Oswald J, and Walter J.
1996. A modified and improved method for
bisulphite based cytosine methylation analysis. Nucleic Acids Res. 24: 5064-6.
2-10 Grunau C, Clark SJ, and Rosenthal
A. 2001. Bisulfite genomic sequencing: systematic investigation of
critical experimental parameters. Nucleic Acids Res
29: E65-5.
2-11 Saha S, Sparks AB, Rago C, Akmaev V, Wang CJ, Vogelstein B, Kinzler
KW, and Velculescu VE. 2002. Using the transcriptome to annotate the
genome. Nat Biotechnol. 20:508-12.
2-12 Brenner S, et al.
2000. Gene expression analysis by massively parallel signature
sequencing (MPSS) on microbead arrays. Nat
Biotechnol.18: 630-4.
2-13 Singer-Sam J. 1994. Quantitation of
specific transcripts by RT-PCR SNuPE assay.
PCR Methods Appl. 3:S48-50.
2-14 Mitra,
R. and Church, G.M. 1999. In situ localized amplification and contact
replication of many individual DNA molecules.
Nucleic Acids Res. 27: 1-6.
2-15 Lizardi, P.M., Huang, X., Zhu, Z.,
Bray-Ward, P.,
2-16 Harrer T, Schwinger
E, and Mennicke K. 2001. A new technique for
cyclic in situ amplification and a
case report about amplification of a single copy gene sequence in human
metaphase chromosomes through PCR-PRINS. Hum Mutat.
17: 131-40.
2-17 Alzahrani AJ, Vallely
PJ, and McMahon RF. 2002. Development of a novel nested in situ PCR-ISH method for detection of
hepatitis C virus RNA in liver tissue. J Virol
Methods 99:53-61.
2-18 Sallstrom JF, Zehbe
I, Alemi M, and Wilander E.
1993. Pitfalls of in
situ polymerase chain reaction (PCR) using direct incorporation of labeled
nucleotides. Anticancer Res. 13:1153.
2-19 Hu, X, Aston C, and
2-20 Ole
2-21 Ole
2-22 Egner A, Jakobs
S, and Hell SW. 2002. Fast 100-nm
resolution three-dimensional microscope reveals structural plasticity of
mitochondria in live yeast. Proc Natl Acad Sci U S A
99:3370-5.
2-23 Lacoste, TD, Michalet, X, Pinaud, F, Chemla, DS, Alivisatos, AP, and S. Weiss. 2000. Ultrahigh-resolution multicolor colocalization of single fluorescent probes. Proc
Natl Acad Sci USA 97: 9461-9466.
2-24 Drummond D.R., Carter N., Cross R.A. 2002. Multiphoton versus confocal high
resolution z-sectioning of enhanced green fluorescent microtubules: increased multiphoton photobleaching within
the focal plane can be compensated using a Pockels
cell and dual widefield detectors. J
Microsc. 206:161-9.
2-25 Maiti, S., Shear, J.B., Williams, R.M., Zipfel,
W., and Webb, W.W. 1997. Measuring Serotonin Distribution in
Live Cells with Three-Photon Excitation.
Science 275: 530-532.
2-26 Xu C, Zipfel W, Shear J.B.,
Williams R.M., and Webb W.W. 1996. Multiphoton
fluorescence excitation: new spectral windows for biological nonlinear
microscopy. Proc Natl Acad Sci
2-27 Sharpe
J., Ahlgren U., Perry P., Hill B., Ross A., Hecksher-Sorensen J., Baldock R.,
and Davidson D. 2002. Optical pro
2-28 Modrek B., and Lee C. 2002. A genomic view of alternative splicing. Nat Genet 30:13-9.
2-29 Kalbfuss B., Mabon
2-30 Link,
A.J., Robison, K. and Church, G.M.
1997. Comparing the predicted and observed properties of proteins encoded in
the genome of Escherichia coli. Electrophoresis 18 (8):1259-1313
2-31 Chen, T.,
Jaffe, J.D.. and Church, G.M.
2001. Algorithms for Identifying Protein Cross-links via
Tandem Mass Spectrometry. Recomb. J Comput Biol. 8:571-583.
2-32 Brizuela L, Braun P, and LaBaer J. 2001. FLEXGene
repository: from sequenced genomes to gene repositories for high-throughput
functional biology and proteomics. Mol
2-33 Yu
D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., and Court D.L. 2000. An efficient recombination system for chromosome engineering in
Escherichia coli. Proc Natl Acad Sci USA 97:5978-83.
2-34 Raymond C.K., Sims E.H., and Olson M.V. 2002.
Linker-mediated recombinational subcloning
of large DNA fragments using yeast. Genome Res. 12:190-7.
2-35 Lin,
Y. and Farmer,
A. 2002. BD Bioscience
Application note. http://www.clontech.com/products/families/creator/index.shtml
2-36 Barsoum J., Brown R., McKee M.,
and Boyce F.M. 1997. Efficient transduction of
mammalian cells by a recombinant baculovirus having
the vesicular stomatitis virus G glycoprotein.
Hum Gene Ther. 8: 2011-8.
2-37 Weng S., Gu K., Hammond P.W., Lohse P., Rise C., Wagner R.W., Wright M.C., and Kuimelis R.G. 2002.
Generating addressable protein microarrays with PROfusion
covalent mRNA-protein fusion technology. Proteomics 2:48-57.
2-38 Roberts R.W. and Szostak J.W. 1997. RNA-peptide fusions for the in vitro
selection of peptides and proteins. Proc Natl Acad Sci
2-39 Forster A.C., Weissbach H., and
Blacklow S.C. 2001. A simplified reconstitution of mRNA-directed peptide
synthesis: activity of the epsilon enhancer and an unnatural amino acid. Anal
2-40 Shimizu
Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T.,
Nishikawa K., and Ueda T. 2001. Cell-free translation reconstituted with
purified components. Nat Biotechnol 19:751-5.
2-41 Beckett,
D., Kovaleva, E., and Schatz, P.J. 1999. A minimal peptide substrate in biotin holoenzyme
synthetase-catalyzed biotinylation.
Protein Science 8: 921-929.
2-42 Yusupov M.M., Yusupova G.Z., Baucom A., Lieberman K., Earnest T.N., Cate
J.H., and Noller H.F. 2001.
2-43 Tanha J., Xu
P., Chen .Z, Ni F., Kaplan H., Narang
2-44 Conrath K.E., Lauwereys
M., Galleni M., Matagne A.,
Frere J.M., Kinne J., Wyns L., and Muyldermans S. 2001. Beta-lactamase
inhibitors derived from single-domain antibody fragments elicited in the camelidae. Antimicrob Agents Chemother. 45: 2807-12.
2-45 Cortez-Retamozo V., Lauwereys M., Hassanzadeh Gh. G., Gobert M., Conrath K., Muyldermans S., De Baetselier P.,
and Revets H. 2002. Efficient tumor targeting by
single-domain antibody fragments of camels. Int J
Cancer 98: 456-62.
2-46 Muruganandam A., Tanha J., Narang S., and Stanimirovic D. 2002. Selection of
phage-displayed llama single-domain antibodies that transmigrate across human
blood-brain barrier endothelium. FASEB J 16: 240-2.
2-47 Hanes J., Schaffitzel C., Knappik A., and Pluckthun A. 2000. Picomolar
affinity antibodies from a fully synthetic naive library selected and evolved
by ribosome display. Nat Biotechnol. 18: 1287-92.
2-48 Shin
S.U., Wu D., Ramanathan R. Pardridge
W.M., and Morrison S.L. 1997. Functional
and pharmacokinetic properties of antibody-avidin
fusion proteins. J Immunol 158: 4797-804.
2-49 Humm A., Fritsche
E., Mann K., Gohl M., and Huber R. 1997.
Recombinant expression and isolation of human L-arginine:glycine amidinotransferase
and identification of its active-site cysteine residue.
2-50 Smith
J., and Modrich P. 1997. Removal of
polymerase-produced mutant sequences from PCR products. Proc Natl Acad Sci
2-51 Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras
K., Gustafsdottir S.M., Ostman
A., and Landegren U.
2002. Protein detection using proximity-dependent DNA ligation assays.
Nat Biotechnol. 20: 473-7.
2-52 Schweitzer
B., Wiltshire S., Lambert J., O'Malley S., Kukanskis K., Zhu Z., Kingsmore
S.F., Lizardi P.M., and Ward D.C. 2000.Inaugural
article: immunoassays with rolling circle DNA amplification: a versatile
platform for ultrasensitive antigen detection. Proc Natl Acad Sci USA 97:
10113-9.
2-53 Truong
K., and Ikura M. 2001. The use of FRET imaging microscopy to detect protein-protein
interactions and protein conformational changes in vivo. Curr Opin Struct
Biol 11: 573-8.
2-54 Singh-Gasson S., Green R.D., Yue Y.,
Nelson C., Blattner F., Sussman M.R., and Cerrina F. 1999. Maskless fabrication of light-directed oligonucleotide microarrays using a
digital micromirror array. Nat Biotechnol.17:
974-8.
2-55 Bulyk,
M.L., Gentalen, E. Lockhart, D.J., and Church, G.M.
1999. Quantifying DNA-protein interactions by double-stranded
DNA arrays. Nature Biotechnology 17: 573-7.
2-56 Willer D.O.,
2-57 Mendel D., Cornish V.W., and Schultz P.G. 1995. Site-directed mutagenesis with an expanded genetic code. Annu Rev Biophys Biomol Struct 24:435-62
2-58 Wang L., Brock A., Herberich B.,
and Schultz P.G. 2001. Expanding the genetic code of
Escherichia coli. Science 292: 498-500.
2-59 Jenuwein T., and Allis C.D. 2001. Translating the histone code.
Science 293:1074-80.