Saeed Tavazoie(1), Jason D. Hughes(1,2), Michael J. Campbell(3), Raymond J. Cho(4) and George M. Church(1)
We present a systematic approach for determining the higher order organization
of the transcriptional regulatory network from whole-genome mRNA expression
data. Time-series of mRNA abundance, measured over two synchronized
Saccharomyces cerevisiae cell cycles, were used to group 3000 genes into
various expression classes which were highly enriched for genes of similar
function. A systematic, upstream DNA motif search identified many
known and putative cis-regulatory elements, highly specific to each expression
class. The identification of many expected cis-regulatory elements,
together with the expression-class specificity of many novel motifs, makes
this combination approach promising for the rapid elucidation of regulatory
network architecture in the myriad of organisms which are now completely
described at the DNA sequence level.
Schematic
overview of our approach
Cluster
members (together with distances and annotations)
Enrichment of clusters for MIPS
functional categories
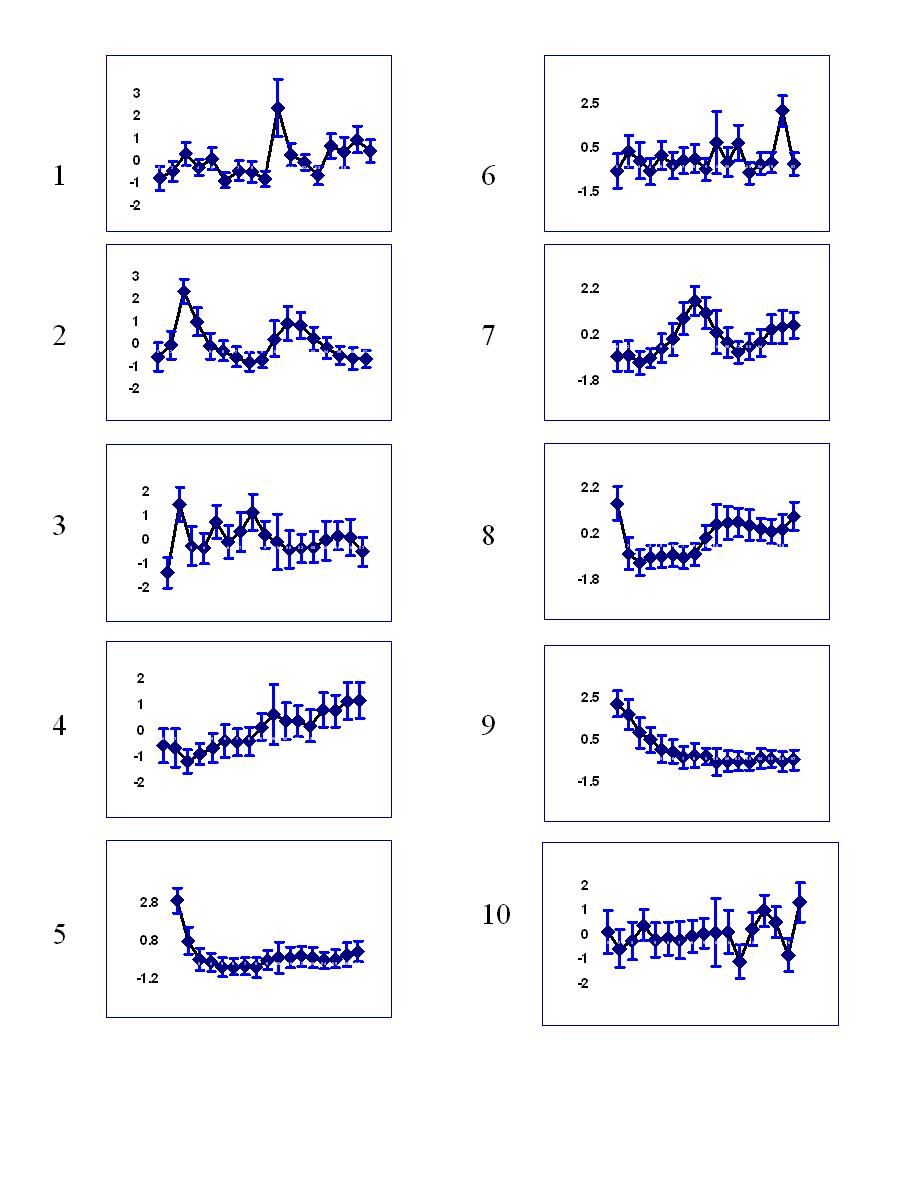
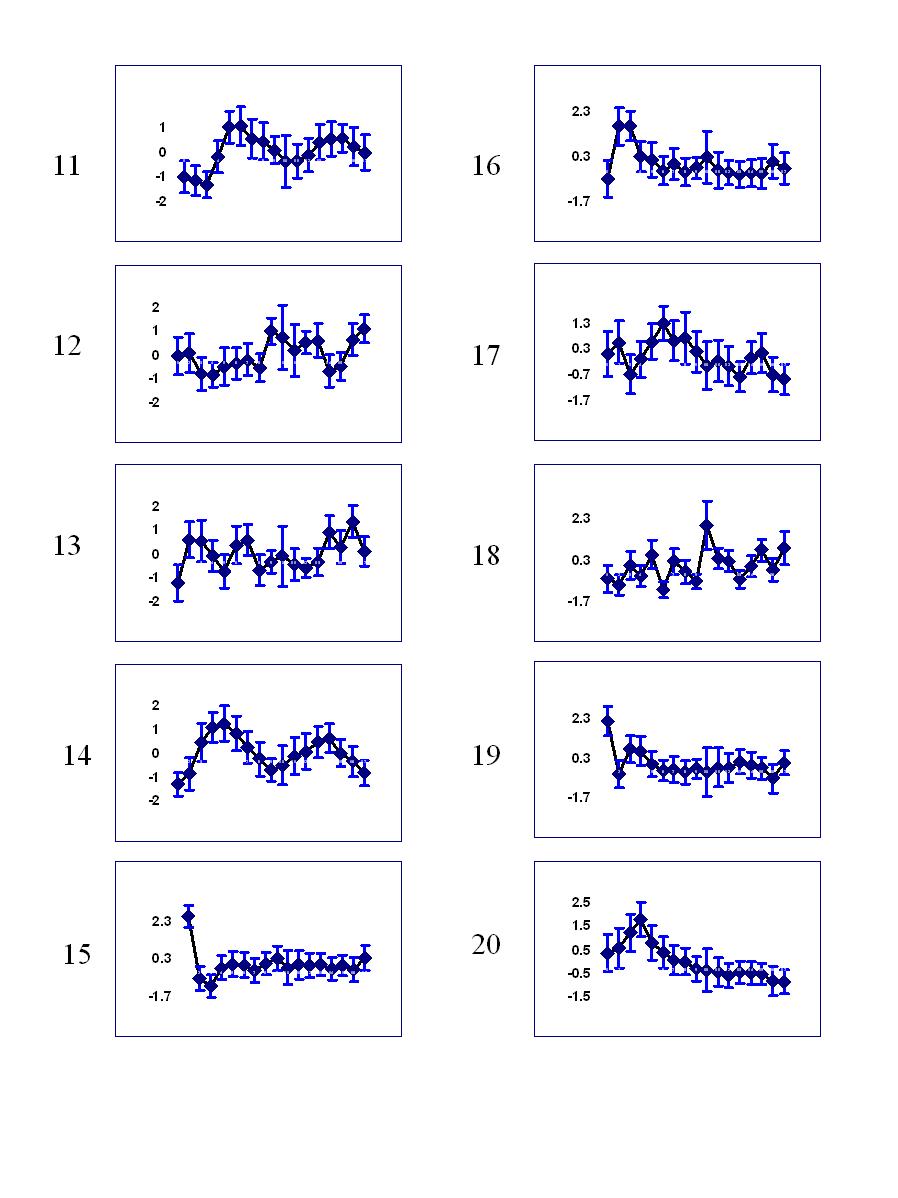
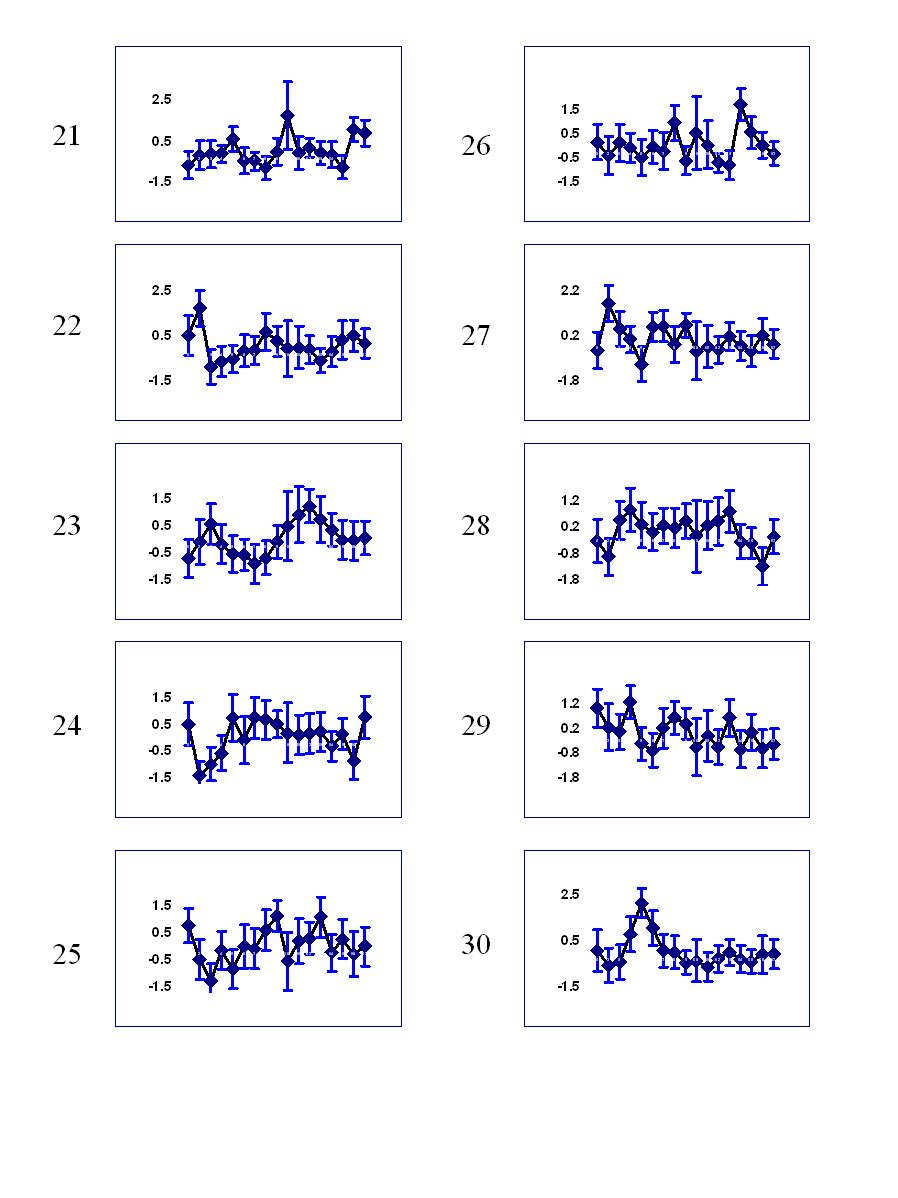
Temporal profile of clusters:
Clusters 1-10
Clusters 11-20
Clusters 21-30
DNA sequence motifs and their statistical characterization
DNA sequence motifs and their
occurences within upstream sequences
{kind=link}
{kind=link}
{kind=link}