March 30, 1998
John Aach
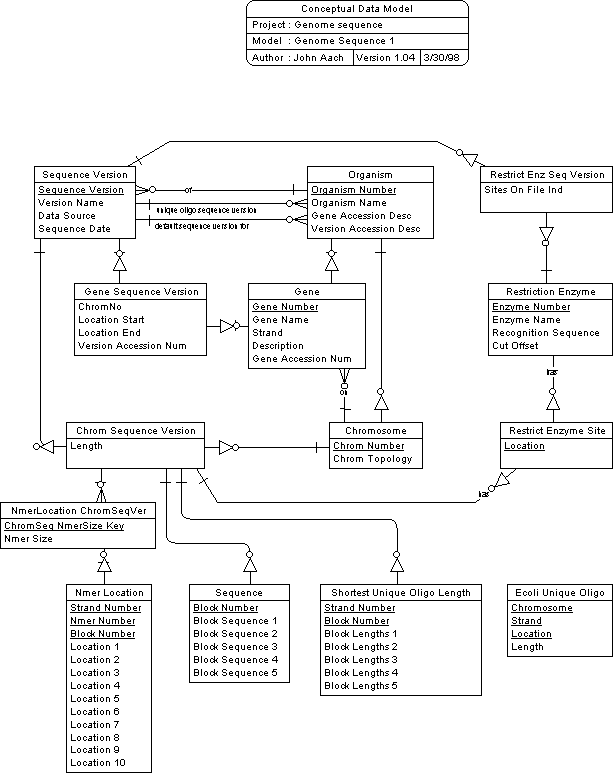
The purpose of the GenomeSequence database is to provide basic storage, retrieval, indexing, and processing of sequence information for wholly sequenced organisms. It provides the following functionality:
Item 3 provided the original motivation for building GenomeSequence. When a new version of a sequence comes out, work done on the basis of earlier sequence versions must be reviewed. For instance, Dereth Phillips discovered several impacts when she compared the first E coli whole-genome sequence to come out of the Blattner lab with the earlier partial sequences she used in the preparation of 46 gene knock-out strains, including:
Discovering the impacts of the revised sequence took several days, partly because the operations of selecting out the relevant gene annotation and sequence data and comparing them across sequences were not automated. To assist in this process, several tables were created on an ad-hoc basis and loaded with annotation and gene location data for the two versions. The ability to use standard database query operations for comparing this data proved helpful and it was decided to create a full database containing not only the annotation but also the sequence data. This would allow the database to be used for other purposes than mere version comparison, which is only of interest for a short period of time when a new sequence becomes available.
A simple application of this first version of GenomeSequence was to provide a flexible and generalized routine for selecting out parts of a genome of interest for purposes of sequence analysis. E.g., it may be desirable to select out upstream regions, intergenic regions, as well as parts of coding regions for selected genes and subject them to homology or motif analysis. Although it is not generally difficult for people to write routines to select out parts of a sequence of interest, the lack of a common, generalized routine forces many people to do essentially the same work over and over again. It would be more efficient to have a common routine. Along similar lines, some aspects of sequence processing are so difficult and resource-intensive that they are difficult to do even once, no less repeatedly. For such it is more efficient to do the processing once and hold the results in a database. It was in this spirit that a unique oligo table was developed for GenomeSequence.
The database was enhanced and a more substantial application built on it with versions 1.03 and 1.04 of the database. This application, PrimerFinder, works off GenomeSequence to select primers for pKO3 knockouts. Among the results of this expansion were the development of an integrated table of shortest unique oligo length data, plus data on restriction enzymes.
Entities in the database are of two basic types - sequence-independent and sequence-dependent. Sequence-independent entities keep track of information that is not expected to change between sequence versions. Organism, Gene, Chromosome, and Restriction Enzyme are of this sort. Examples of the kinds of information that are kept in these entities include version-independent gene accession numbers, such as the "b" labels introduced and maintained by the Blattner lab for E coli genes, and chromosome topology. Sequence-dependent entities contain information that is expected to change between sequence versions. Gene Sequence Version, Chrom Sequence Version, Restrict Enzyme Site, Nmer Location, and, of course, Sequence itself are of this sort. Examples of sequence-dependent information kept in these entities include sequence-dependent gene accession numbers (e.g. GenBank PIDs), gene coding region locations in a sequence, chromosome length, and the locations of restriction sites.
The principal way these entities are related is to make sequence-dependent entities dependent on their sequence-independent counterparts along with the Sequence Version entity, which records a version of a sequence. Thus, Gene Sequence Version is dependent on both Gene and Sequence Version, and Chrom Sequence Version is dependent on Chromosome and Sequence Version. Sequence is itself dependent on Chrom Sequence Version, and Restrict Enzyme Site is dependent on Restriction Enzyme and Chrom Sequence Version
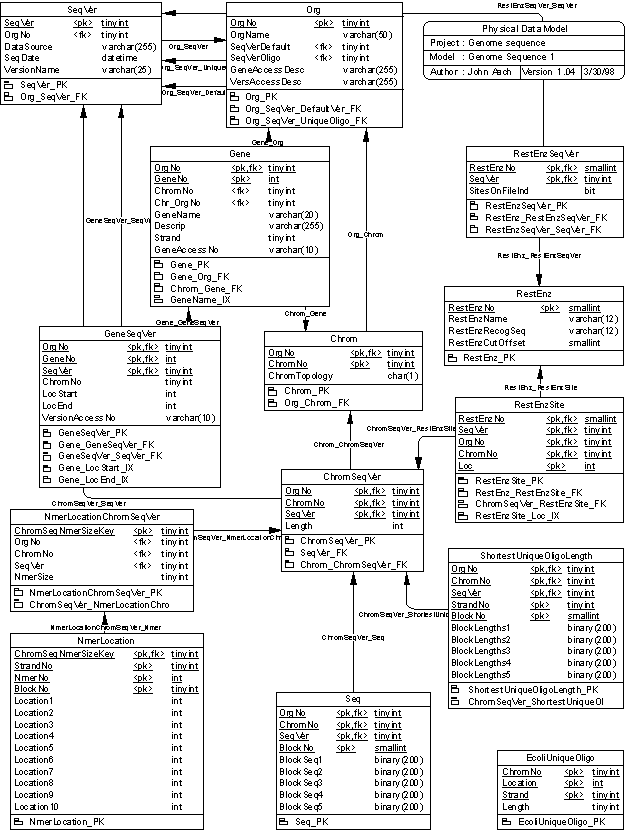
Sequence data on the database is maintained in table rows that contain 1000 bytes of bitstring data (specifically, each row of the Sequence table contains five 200 byte binary strings). Nucleotides are represented by 2 bits, with 'a' = 00, 'c' = 01, 'g' = 10, and 't' by 11 (numbers are binary). A single row of the Sequence table thus contains information on a 4000 base segment of a chromosome. Each such row is termed a 'block' and is keyed by Block Number in addition to Organism Number, Chromosome Number and Sequence Version.
Three consequences of this arrangement are (a) sequences stored on GenomeSequence must be entirely unambiguously called, (b) proper processing of sequence data must take into account sequence lengths as well as sequence data to prevent binary 00s padding the end of a sequence block from being interpreted as extra 'a's, and (c) built-in Sybase pattern-matching for bitstrings (using the "like" operand) cannot be effectively used. The effect of (b) and (c) together is that analysis of sequence data from the database will generally require programming and not native SQL operations.
That the database has been built with these characteristics is the result of a design decision. In particular, (a) it was thought that the functional genomics work this database will support will generally require fully-sequenced organisms and prefer fully-called sequences, anyway. As for (b) and (c), it was judged that the native SQL tools offered by relational databases cannot provide much help towards standard sequence analysis tasks anyway, so that little advantage is gained by designs that enable their use.
Only one strand of DNA sequence is kept on the database - the strand that is featured in the source genome sequence file. This is designated as strand 1.
Sequence locations are maintained in GenomeSequence for gene coding region boundaries and unique oligos. Two conventions apply to handling of locations:
In line with the thought that the only time it will be useful for multiple versions of a sequence to be maintained is when a new version has come out and needs to be compared with the old one, one version of a sequence may be designated as a "default" for an organism through a relationship between Organism and Sequence Version. This will indicate the version that the database and associated procedures will use by default, unless another version is specifically indicated. It is anticipated that when a new version of a sequence is first loaded, the old version will be the default until most of the work of comparing sequences is done. Afterwards, the new version will be made the default and the old version maintained for a time to allow 'mop up' comparisons and analysis to be done.
Version 1.03 of GenomeSequence maintains two tables concerned with unique oligo data in the genome. The original version of GenomeSequence featured an E coli-specific table of unique oligos from that was not fully integrated with the database. With the PrimerFinder application, a fully integrated approach was developed that maintains a different kind of unique oligo data.
While both the old and the new tables are maintained in version 1.03, the old version is slated to be abandoned over time. It has been maintained only because query applications have been written based on it that are available on the GenomeSequence web interface. These applications could be rewritten using the new unique oligo table, but as this step has not yet been taken it is easier for the moment to maintain the old one as well. Both versions will be described here, starting with the new one.
The version of unique oligo data new with GenomeSequence 1.03 is the Shortest Unique Oligo Length table. The data it maintains is the length of the shortest unique oligo that starts at any 5' location in the genome. Its structure is similar to Sequence data in that unique oligo data is stored in 1000 byte blocks that cover a range of sequential locations in the genome. In this case, one byte of unique oligo data is maintained for every 5' location (vs two bits in Sequence). Thus, no shortest unique oligo length > 255 bases may be maintained on the database, and locations with longer shortest unique oligo lengths get capped at 255. Like Sequence, the 1000 bytes are broken up into five 200 byte columns, with each column defined as a bitstring. (Unlike Sequence, the data is being stored in bitstring form not because it must be analyzed at the bit level, but to make management of load files easier. If the data were stored in characters, the fact that each character represented a numeric length would result in many special characters that would require careful handling and which, not being displayable, would be harder to debug.)
Shortest Unique Oligo Length is defined as dependent on Chrom Sequence Version, thereby making it sequence version-dependent and providing Organism Number and Chromosome Number as parts of the primary key. Strand Number and Block Number fill out the remainder of the primary key and complete the apparatus needed to relate records to locations in the genome. As with the Sequence table, Block Number starts at 1 and increments for each 1000 bases in the genome.
Unlike Sequence, Shortest Unique Oligo Length maintains information on both strands. This is because the shortest unique oligo lengths that start from 5' locations on strand 2 cannot be easily derived from the corresponding values for the 5' strand 1 locations. The relationship between strand 1 and 2 values is rather more subtle: Let the Shortest Unique Oligo Length value for a particular chromosome location on strand 1 be L1, and that for the same location on strand 2 be L2. Then L2 actually represents the length of the shortest unique oligo on strand 1 that *ends* at that location. This relationship is used in the PrimerFinder system to assess the 3' end uniqueness of chromosomal segments of primers.
Shortest Unique Oligo Length information is derived from the output of a suffix array program that is a modified version of one originally written by Tim Chen and run with his assistance. Tim Chen was also the person who originally suggested a blocked structure for storing this data. The program puts out a file of shortest unique oligo lengths on both strands of all chromosomes, along with their locations, with unique oligo lengths capped at 255. (At several bytes for every strand/location in the chromosome, this is a huge file.) The data is then sorted by location and converted into the blocked bitstring format described above. The suffix array program contains logic to handle wraparound for circular chromosomes that is under parameter control.
This is the original version of unique oligo information in GenomeSequence. As already described, Ecoli Unique Oligo was implemented as an isolated table without relationships with the rest of the database. It consists of the locations and lengths of 'minimal' unique oligos in the E coli chromosome with lengths between two fixed bounds (7 to 11). Locations were treated according to the conventions in 3 above where the unique oligo is treated as a segment of DNA, i.e., the location always represents the left-most endpoint of the oligo regardless of what strand it is on. Thus, use of length and strand is required to determine the other endpoint of the oligo.
The design of the Ecoli Unique Oligo table required one record for each unique oligo in the genome within the given size range. It was because this requires a huge table that only a small size range of oligos was accommodated, a direction reinforced by the thought that only the shortest unique oligos in the genome would be of interest. (The table as it stands today (2/18/98) based on the M52 version of the E coli sequence has 904776 rows.) The huge size also led to the isolation of the table from the rest of the database, for to integrate it with other tables would require incorporation of foreign keys into Ecoli Unique Oligo that would substantially increase storage requirements. The benefits of integration also appeared unclear. Not only is it hard to imagine a useful query that would process both E coli and (e.g.) yeast unique oligos, but the presence of both in the same table could impact performance as queries on E coli data could force navigation around yeast data and vice versa.
Instead, Ecoli Unique Oligo was designed with a structure that could be 'cloned' and applied to other organisms, allowing common routines to be applied to all. For this reason it maintained a Chromosome Number, which might be needed by other organisms even if not E coli. It was conceived that unique oligo tables would be created only for organisms that needed them, and only for one sequence version at a time. The sequence version was maintained on the database through a relationship between Organism and SequenceVersion.
Shortest unique oligos in E coli were determined by an earlier modified version of Tim Chen's suffix array program. In this case, the program was equipped with a size filter and only reported on oligo locations and lengths within the given size range. To go from shortest unique oligo lengths to 'minimal' oligos required a pass of the data through another series of programs. A fact about unique oligos is that any oligo containing them is also unique. The suffix array program effectively looks 3'-wards of any 5' starting location until it finds a unique oligo, and thus ensures that the unique oligos it discovers are not unique by virtue of extending 3'-wards from a shorter unique oligo. But these oligos may yet be unique by virtue of starting 5'-wards of a shorter unique oligo. The signature of such a situation is that the suffix array results will show a series of unique oligos at consecutive locations, each one base shorter than the previous one. The programs that convert suffix array data to minimal unique oligo look for this situation and eliminate all but the shortest unique oligo records.
This processing has not been done on the newer Shortest Unique Oligo Length table data, which does not report minimal unique oligos. However, minimal unique oligos can be derived from Shortest Unique Oligo Length data by the following simple logic: If one starts at a given 5' location and goes to the end of the shortest unique oligo beginning there according to the table, and then take the shortest unique oligo lengh for that end location *on the opposite strand* and go back that amount, one will have found a minimal unique oligo. In fact, this will be the minimal unique oligo by virtue of which, by 5'-wards extension, the oligo indicated for the original 5' location is unique. Implementation of this logic will allow the current GenomeSequence web query application for unique oligos to be based on Shortest Unique Oligo Length, and allow elimination of Ecoli Unique Oligo.
A PrimerFinder application enhancement to take genomic PCR noise into account in primer selection drove the creation of two new tables in GenomeSequence version 1.04 to maintain Nmer Location data. The term "genomic PCR noise" refers to existence of sites in the genome other than intended primer sites to which the 3' ends of primers may hybridize that are sufficiently close and in the right orientation to generate PCR products. The purpose of the Nmer Location table is simply to hold, for any given Nmer of a specific size, all the locations of the Nmer in the genome. In its initial implementation, Nmer Location has been loaded with the locations of all 9mers in the E coli K-12 MG1655 genome, M52 sequence version (referred to as the "reference genome" below).
Conceptually, Nmer Location stores a list of chromosome locations for an Nmer of a given size in a given sequence version. Separate lists are stored for each strand of the genome as strands must be distinguished in primer selection logic. The length of the location list is variable, ranging from 1 to 293 in the reference genome (average = ~17.7 occurrences). Note that Nmers with no occurrences are simply not registered in the table; there are 5595 of these in the reference genome. The total number of locations recorded is twice the size of the genome (once for each strand), distributed over a maximum of 4**n Nmers of size n.
To handle this variously sized list of locations, Nmer Location rows maintain locations in blocks of 10. A key field BlockNo is provided to support multiple rows for a given Nmer in a genome. Locations are stored in order and any available location columns in the last block are given 0 values. Location columns are implemented as integer fields. 1169424 rows were required to store 9mer locations for the reference genome.
In addition to BlockNo Nmer Location requires key fields that map to OrgNo, ChromNo, SeqVer, StrandNo, and Nmer. A number of steps have been taken to reduce the amount of storage and other database overhead required to manage this large set of key fields. (1) Nmers are stored not as character strings representing Nmer sequences but as integer values that correspond to them. The database name of this column is NmerNo. The translation is simply to treat each Nmer sequence as a base 4 integer where 'a' = 0, c = '1', g = '2', and t = '3'. Note that this means that knowledge of the size of the Nmer (Nmer Size) is required to translate an NmerNo back to an actual sequence. (2) As the values OrgNo, ChromNo, SeqVer, and Nmer Size will be constant over a given genome and sequence version, each combination of these for which Nmer Locations are maintained is associated with a unique one byte integer key in the separate table NmerLocationChromSeqVer. The name of this one byte key is ChromSeqNmerSizeKey. This eliminates the need for these multiple values to be embedded in Nmer Location rows themselves. (3) Nmer Location indexes relating ChromSeqNmerSizeKey back to individual NmerLocationChromSeqVer records have not been generated.
With these design features, Nmer Location keys are reduced to ChromSeqNmerSizeKey, StrandNo, NmerNo, and BlockNo.
This came in with version 1.03 of the database. Locations of all sites of selected restriction enzymes are now recorded in a Restrict Enzyme Site table. Dependency relationships between this table and its parents Restriction Enzyme and Chrom Sequence Version allow restriction sites to be recorded for different versions of an organism sequence. Another table, Restrict Enz Seq Version, is used to register the fact that a set of sites on Restrict Enzyme Site has been included for a given enzyme and sequence version. This has been included to disambiguate potential situations in which a query on Restrict Enzyme Site has returned no sites: If the Sites On File indicator of the Restrict Enz Seq Version row corresponding to the query is on, the result-less query can be interpreted as indicating that there are, in fact, no sites satisfying the conditions of the query. Without such an indicator, it would not be known whether there were no sites or whether sites for the enzyme and sequence version under consideration were simply never recorded on the database.
For performance reasons, ChromNo from Gene has been denormalized and is maintained also on GeneSeqVer, and indexes have been set up in the physical model on GeneSeqVer that include ChromNo and LocStart, and ChromNo and LocEnd.
This data model was prepared in PowerDesignor 6.1. This document was produced via the PowerDesignor report writer and edited in Microsoft Word 97 as HTML.
Please contact John Aach in case of problems or suggestions:
Copyright (c) 1998 by John Aach and the President and Fellows of Harvard University