Wobble Sequencing [JAS,
Description of Wobble Sequencing
Proof of Principle on Loaded Beads (T1..T5)
Proof of Principle on Emulsion Beads (T1..T5)
Description
of Wobble Sequencing
Although this is still a
method based on sequencing by polymerase-extension, it differs from FISSEQ and pyrosequencing in that base-additions are not
“progressive”. Instead, after a given
single-base-extension (SBE), the sequencing primer is stripped from the
bead-immobilized templates and a new primer is hybridized. The trick to getting beyond the first base is
that each sequencing primer in the set “reaches” out to a defined position in
the unknown unique sequence of the template.
A sequencing primer, from 5’ to 3’, thus consists of an “anchor
sequence” that is complementary to the constant sequence on the template [identical
to the sequencing primers that we’ve been using but a bit shorter to aid in
primer-strpiping], and a defined number of additional
“universal” bases, that will hybridize to the unknown sequence regardless of
what it is. If, for example, there are
three fixed universal bases, then the sequencing primer is positioned to sequence
the fourth base via SBE with labeled nucleotides. After a single-base-extension and data acquisition,
extended and unextended primers are stripped (with
heat) and a new primer is annealed that has a different number of universal
bases, thus querying a different base-position within the unknown
sequence. In this simplest iteration of
the scheme, one only needs a set of N primers to achieve a read-length of N.
The primary challenge is how
to define the universal bases. Universal
bases are synthetic nucleotide analogs that ideally pair with equal affinities
to each of the natural nucleotides, and are readily accepted as substrates by
natural enzymes. Examples include
5-nitroindole, 3-nitropyrole, deoxyinosine, etc. Another possibility would be to use natural
bases, but order the sequencing primer oligonucleotides with a full “wobble” at
universal positions, such that all possible sequencing primers (or some random
subset of all possibilities) is present during
hybridization. I tried all of the above
with marginal success.
The failure is pretty easy
to explain in retrospect by the facts that (1) regardless of how they behave in
hybridization, polymerases tend not to like to extend off of unnatural bases;
(2) for natural nucleotides, matching at the 3’-terminal of the sequencing
primer is the most critical for efficient primer extension, but the terminal
bases of any sequencing primer will make the least contribution to hybridization
kinetics (relative to other “wobble” positions). The result is that even if some “sorting”
goes on with “wobble” sequencing primers during primer hybridization, the
primers that ultimately hybridize are quite likely to be mismatched at the 3’
terminus.
The approach that finally started
to show some success involved fixing the terminal two bases of a given
sequencing primer, but allowing the remainder of bases at “universal” positions
to be synthesized with a full “wobble” of natural bases. The disadvantage of this compromise is that
16 separate hybridizations are required for each “reach” length (4^2
combinations of the two terminal bases).
This is mitigated by the fact that polymerases don’t extend off mispaired termini very well at all, so a given extension
set reveals the identity of both the two terminal bases and the extended base. So the average efficiency of the process is 3/16
= 0.188 bases per cycle.
Non-terminator FISSEQ, by comparison,
yields approximately ~0.50
bases-per-cycle (assuming no homopolymer resolution and thus counting multi-base
runs as single extensions; someone double-check this if you have a second). By this consideration, achieving an identical
read-length will require ~2.67 times as many cycles in the 2bp-matched-wobble-sequencing
system.
Regardless, if we or someone
else eventually developed a four-color reversible-terminator FISSEQ system, there
would be no real advantage to the wobble-sequencing method as it would provide
the same information (i.e. homopolymer resolution) but require 5 times as much
work.
Summary of Advantages
1.
First
and foremost, provides a quick solution to the homopolymer problem.
2.
Mistakes
(manual) and inefficiencies (biochemical) are non-cumulative.
3.
Technology
development for longer reads is greatly expedited (i.e. don’t have to cycle out
to test a hypothesis for improving read-lengths)
4.
Better
signal than current FISSEQ system (in which a desire for signal has to be
balanced against a desire to minimize the fraction of extended templates with
cleaved linker as it inhibits the polymerase).
5.
Lack
of requirement to take extensions to completion opens up polymerase-choice (and
amounts).
Summary of Disadvantages
1.
The
“terminally-matched” approach (i.e. xN.2 primers) requires ~3 times as many
cycles than FISSEQ to achieve the same read-lengths.
2.
Probably
a lower prospect of ever achieving read-lengths > 20.
3.
Has
only been tried on a small number of templates
Arguments against the listed
disadvantages are that (1) if it works, this is no biggie, (2) read-lengths of
10-20 will be sufficient for most purposes, especially in the context of a
paired-strategy, and (3) the same could be said of FISSEQ.
We also might imagine that
the overall efficiency could by improved by some enzyme engineering for greater
permissiveness with respect to mismatches (e.g. the M1/M4 variants of Taq) or alterations to the primer design strategy.
In the current protocol, a
typical cycle is as follows:
1. Hybridize sequencing primer (15
minutes, 10 uM primer in 6x SSPE, 40-50’C)
2. Extend (4 minutes, SSB + polymerase +
nucleotide)
3. Wash (2 minute)
4. Image acquisition
5. Strip primer (5 minutes,
A couple notes about the
above:
(a) If the wobble-bases are
fixed (poly-A, poly-G, poly-C, or poly-T instead of poly-N), extensions are no
longer efficient. This implies that that
some degree of “sorting” is going on during the hybridization that is critical
to the overall process working. Hoping
for this to occur, the “anchor sequence” is purposefully short (Tm = 37’C if it
were alone), weighting the hybridization process to depend to a greater degree
on the wobble-sequences.
(b) Initial data suggests
that Sequenase is significantly better than Klenow for this approach.
(c) Primer-stripping was
initially very inefficient with beads.
It only started working when
we started top-layering the beads (not too surprising,
in retrospect).
Primer Nomeclature
So
a typical primer-name below is “37C.2N.CA”.
For all of the primers below, the anchor sequence is a trimmed version
of the original FISSEQ primer for the T1..T5 template. The “37C” (or “23C” or whatever it is)
indicates the extent to which it has been trimmed (i.e. 37C is the Tm of the
anchor sequence if it were a stand-alone primer). The “2N” indicates that the anchor-sequence
is followed by two full wobble bases, and the CA indicates the fixed two
terminal bases. This primer would extend
to the 5th base, thus sequencing 3 bases (base 3, 4 and 5) on 1/16th
of the templates of a random library.
In
the experiments described below, we have focused on primers with even numbers
of wobble-bases and terminal bases that match at least one of the five T1..T5 templates (so that at least something will
extend). For a given reach-length, this
is approximately 1/4th of the primers that would be required in a
real sequencing experiment involving sequencing of genomic fragments. However, we’re also being slightly
conservative in that one could do multiples of three for the number of
wobble-bases, rather than multiples of two.
We have some redundancy built in here as this is just a
proof-of-principle experiment. For
example, 37C.2N.XX sequences bases 3, 4 and 5.
37C.4N.XX sequences bases 5, 6 and 7.
Base 5 is sequenced twice (as is base 7, base 9, etc.)
Proof of
Principle on Loaded Beads (T1..T5) [Slide EZ]
Slide “EZ” has top-layered,
1 uM beads with loaded T1..T5
templates. As mentioned above, I
only tried a subset of the primers that would be required in a full sequencing
experiment on unknown sequence. I ordered
primers to sequence through to the 11th base on all five templates
(37C.0N.XX through 37C.8N.XX). I also
ordered just one primer for 37C.10N.XX through 37C.18N.XX (as initial data
suggested that we’d be pushing it to go beyond ~10 bases with this method). Details are as follows. See commentary after for more precise
description of the fields here. TR =
No Primer Name Primer
Seq Hyb Extend With Templates AS NAS A:N Ratio
1 37C.0N.CA CCTCATTCTCT
CA 42c 35’ Cy3-C T1(+T4) 0.87 0.06 15.3
2 Cy5-G
T4 0.27 0.02 12.3
3 TR-G T4(+T1) 1.15 0.06 19.4
4 37C.0N.GT CCTCATTCTCT
GT 42c 45’ TR-G T2 0.83 0.04 19.5
5 37C.0N.AT CCTCATTCTCT
AT 42c 20’ TR-G T5 0.21 0.07 3.0
6 37C.0N.AG CCTCATTCTCT
AG 42c 25’ Cy3-T T3 0.64 0.05 13.7
7 37C.2N.CA CCTCATTCTCT
NN CA 42c 20’ Cy3-C T1 2.16 0.06 38.9
8 37C.2N.GT CCTCATTCTCT
NN GT 42c 60’ TR-G T2,T5 0.35 0.04 8.7
9 37C.2N.TG CCTCATTCTCT
NN TG 42c
20’ Cy3-C T3 1.06 0.06 16.9
10 37C.2N.GC CCTCATTCTCT
NN GC 42c 20’ Cy3-C T4 1.59 0.07 22.7
11 37C.4N.CA CCTCATTCTCT
NNNN CA 45c 40’ Cy3-C T1 1.72 0.06 27.0
12 37C.4N.GT CCTCATTCTCT
NNNN GT 45c
27’ TR-G T2 0.52 0.16 3.2
13 37C.4N.CT
14 37C.4N.GA CCTCATTCTCT
NNNN GA 45c
15’ TR-G T5 0.27 0.04 7.0
15 37C.4N.CG CCTCATTCTCT
NNNN CG 45c
15’ Cy3-A T4 2.53 0.12 20.5
16 37C.6N.CA CCTCATTCTCT
NNNNNN CA 50c 15’ Cy3-C T1,T3 0.53 0.10 5.3
17 37C.6N.GT CCTCATTCTCT NNNNNN GT 50c 15’ TR-G T2 0.12 0.18 0.7
18 37C.6N.AA CCTCATTCTCT
NNNNNN AA 50c 15’ Cy3-C T4 0.86 0.09 9.2
19 37C.6N.GA CCTCATTCTCT
NNNNNN GA 50c 40’ Cy5-G T5 0.07 0.02 3.1
20 37C.8N.CA CCTCATTCTCT
NNNNNNNN CA 50c 30’ Cy3-C T1,T3 0.35 0.10 3.5
21 37C.8N.GT CCTCATTCTCT
NNNNNNNN GT 42c 15’ Cy5-G T2 0.31 0.04 7.0
22 37C.8N.CG CCTCATTCTCT
NNNNNNNN CG 42c 30’ Cy3-A T4 0.75 0.09 8.1
23 37C.8N.GC CCTCATTCTCT
NNNNNNNN GC 42c 15’ Cy3-T T5 0.34 0.06 5.9
24 37C.6N.GT CCTCATTCTCT
NNNNNN GT 40c 15’ Cy5-G T2 0.35 0.04 8.3
25 37C.6N.GA CCTCATTCTCT
NNNNNN GA 40c 15’ Cy5-G T5 0.07 0.02 2.9
26 23C.10N.AC TCATTCTCT NNNNNNNNNN AC 30c
19’ Cy3-C T4 0.14 0.07 2.1
27 37C.12N.CG CCTCATTCTCT
NNNNNNNNNNNN CG 37c 18’ Cy3-A T4 0.74 0.10 7.4
28 Cy5-T T3 0.03 0.06 0.6
29 37C.14N.AT CCTCATTCTCT NNNNNNNNNNNNNN AT 37c 15’ Cy3-C T4 0.08 0.08 1.0
30 37C.16N.CC CCTCATTCTCT NNNNNNNNNNNNNNNN CC 50c 15’ Cy5-A T1,T2,T4,T5 0.11 0.16 0.7
31 37C.18N.CC CCTCATTCTCT NNNNNNNNNNNNNNNNNN CC 50c 15’ Cy5-A T3 0.17 0.10 1.7
32 (Sequencing Primer, Cy3 labeled)
33 (Post-strip, Cy3 signal)
34 (Post-strip, Cy5 signal)
35 (WL)
The first column of numbers
indicates the cycle number assigned to a given query. The second and third columns indicate the
sequencing primer used, and the fourth column indicates the conditions of
hybridization. The fifth column
indicates the base(s) used to extend, and the 6th column indicates the
templates expected to add. The remaining
columns indicate the best-fit slope coefficient for adders and non-adders, and
finally the ratio of these values. Note that this is useful, but not the perfect statistic. Calls about which cycles worked and which didn’
Failures are listed in
yellow. The first failure (cycle 17), was
probably due to manual error in preparing the extension mix, as its repeat
(cycle 24) worked quite well, and this primer worked well in the emulsion-bead
experiment below as well. The remaining
failures obviously seem to correlate with attempts at longer reads. The 37C.12N.CG primer, interestingly, works
quite well for one template but not another.
In a subsequent experiment, using Sequenase
instead of Klenow resulted in both templates working
with this primer. This was the main
reason that I used Sequenase in the below experiment
(on emulsion-beads). Sequenase
also seems to yield greater signal in general than Klenow
in this protocol.
There appear to be some
other trends: (a) poor performance of “G” extensions, not unexpectedly; this
gets better with Sequenase also; and (b) poor
performance of the T5 template in terms of signal yield at any given cycle when
it is expected to extend. This is a
little harder to explain but I have some ideas.
It doesn’t normally behave like this, so I’m guessing it has to do with
the shortening of the anchor of the sequencing primer (some other experiments
hint at this mattering). Other data suggests
that the size of the anchor may not be so important, so there might be a
solution there.
The good news is that we
obtain ~11 base-pair reads on all five templates, and all observations appear
consistent with expectation. A 15-bp
read is obtained on one of the templates (T4), but results are not consistent
(i.e. cycle 28) and we experience failure beyond base 15 (cycles 29-31).
Blue, in the below,
indicates bases that were ultimately sequenced in the above experiment; yellow
indicates bases attempted and failed, and the remainder were not attempted:
T1 CACACACACACACACACTCCACCACT
T2 GTGTGTGTGTGTGTGTCCACCACTCT
T3 AGTGCTCACACACGTGATCCACCACT
T4 CAGCCGAACGACCGATCCACCACTCT
T5 ATGTGAGAGCTGTCGTCCACCACTCT
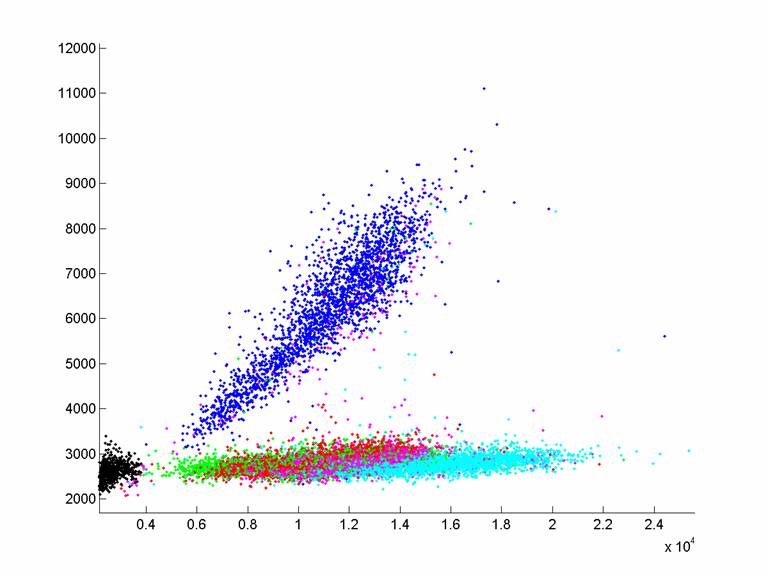
Sample graph (extension with
37C.8N.CG, sequencing bases 10,11,12 on T4)
Proof of
Principle on Emulsion Beads (T1..T5) [Slide G2]
Since the above worked so
well, we decided to try it immediately on emulsion-generated beads (again, just
the T1..T5 templates).
This is Slide G2, using beads prepared by Greg on
On a side-note, Greg has
been optimizing the emulsion-protocol for the human-genomic library, but it
looks like the optimizations were such that the signal on the
emulsion-generated T1..T5 beads are literally through
the roof. The FISSEQ primers have
historically worked better than the human-genomic library primers so this is
not terribly surprising in retrospect, but still really nice. This also suggests that we should try to get
these primer sequences flanking whatever libraries we make in the future, or do
a bake-off of primer sequences and find a set that is even more suited for
emulsion PCR.
Greg diluted these templates
independently, only mixing them as they went into the emulsion mix. The reason for this is that they are
single-stranded, and this procedure minimizes their binding to one another,
which confounds results. However, the
ratios of the five templates clearly deviate from 1:1. The initial set of primers
used on these templates were the 37C.0N.XX series, which essentially
establishes the identity of each bead. As
the fraction of beads with 1+ template was high, it was not surprising that we
observed a high fraction of non-clonal beads.
We were only imaging approximately 1% of the gel (25 frames) at each
cycle. The overall numbers were as
follows:
No template 29,658
Weakly amplified 10,164
Strong clonal 13,350
T1 = 57
T2 = 8,945
T3 = 2,165
T4 = 1,834
T5 = 349
Strong non-clonal 7,668
Total 60,840
The numbers are generally
consistent with what one would expect from Poisson statistics, but with a
modest excess of non-clonal beads. It
seems likely that some of modest fraction of the “no template” beads actually
don’t participate in the distribution (e.g. are excluded b/c they are in the oil
compartment, or in a compartment that is too small to initiate PCR, etc.)
The initial analysis of clonality and identity, which was based on the 37C.0N.XX
primers, led me to focus only on primers that extended either
T2, T3, or T4, as these dominate the slide. Relative to the above there are also changes
to the hybridization conditions and modified nucleotides, but the most
important difference (other than the fact that these are emulsion-generated
beads) is that we are using Sequenase instead of Klenow.
Primer
Name Primer Seq Hyb Extend With Templates Add Slope Non-add Slope A:N Ratio
1 37C.0N.CA CCTCATTCTCT
CA 42c 15’ Cy3-C T1 0.69 0.009 80.8
2 Cy5-A T4 0.33 0.004 87.2
3 37C.0N.GT CCTCATTCTCT
GT 42c 15’ Cy5-G T2 0.19 0.006 29.1
4 37C.0N.AG CCTCATTCTCT
AG 42c 15’ Cy5-T T3 0.78 0.02 44.5
5 37C.0N.AT CCTCATTCTCT
AT 42c 15’ Cy5-G T5 0.04 0.006 6.4
6 37C.2N.GT CCTCATTCTCT
NN GT 42c 50’ Cy5-G T2,T5 0.20 0.05 4.1
7 37C.2N.TG CCTCATTCTCT
NN TG 42c
15’ Cy3-C T3 0.28 0.01 25.6
8 37C.2N.GC CCTCATTCTCT
NN GC 42c 15’ Cy3-C T4 0.67 0.01 46.2
9 37C.4N.GT CCTCATTCTCT
NNNN GT 45c
15’ Cy5-G T2 0.14 0.01 14.4
10 37C.4N.CT
11 37C.4N.CG CCTCATTCTCT
NNNN CG 45c
15’ Cy3-A T4 0.86 0.02 40.6
12 37C.6N.GT CCTCATTCTCT
NNNNNN GT 47c 15’ Cy3-G T2 0.17 0.01 12.8
13 37C.6N.CA CCTCATTCTCT
NNNNNN CA 47c 15’ Cy3-C T1,T3 0.45 0.03 17.3
14 37C.6N.AA CCTCATTCTCT
NNNNNN AA 47c 15’ Cy3-C T4 0.61 0.02 29.2
15 37C.8N.GT CCTCATTCTCT
NNNNNNNN GT 50c 30’ Cy5-G T2 0.11 0.01 9.5
16 37C.8N.CA CCTCATTCTCT
NNNNNNNN CA 50c 15’ Cy3-C T1,T3 0.27 0.02 12.1
17 37C.8N.CG CCTCATTCTCT
NNNNNNNN CG 50c 15’ Cy3-A T4 0.27 0.03 10.2
18 23C.10N.AC TCATTCTCT NNNNNNNNNN AC 37c
30’ Cy3-C T4 0.27 0.02 11.0
19 37C.12N.CG CCTCATTCTCT
NNNNNNNNNNNN CG 54c 18’ Cy5-T T3 0.07 0.04 1.7
20 Cy3-A T4 0.03 0.03 1.4
21 37C.14N.AT CCTCATTCTCT NNNNNNNNNNNNNN AT 54c 15’ Cy3-C T4 0.09 0.03 3.0
22 37C.12N.CG CCTCATTCTCT
NNNNNNNNNNNN CG 54c 18’ Cy5-T T3 0.05 0.03 1.9
23 Cy3-A T4 0.22 0.02 11.6
Note: On cycle 19/20, as
listed above, I accidentally stripped before reading the Cy3 signal out. Interestingly (and assuming that it worked,
which data from Slide EX suggests it should have), less than 30 seconds in
Note that the calls about
what worked and what didn’t work are based on visual inspection of the
graphs. Thus, even though
37.12N.CG->T has lower “ratios” than 37C.14N.AT->C, it still appears to
have worked, whereas 37C.14N.AT->C appears to have not worked.
I stripped this slide and
re-annealed sequencing primer at the conclusion to see what extent the
templates had fallen off due to heat-exposure, etc. The difference between the two sets of images
(pre-sequencing and post-sequencing) was essentially negligible. It’s actually startling how consistent they
were with one another, and indicates that we are not losing template over the
course of the experiment. This
inspection also demonstrates quite clearly that the extent of gel warping over
the ~20 cycles was negligible (a concern, with the heat treatments, etc.)
This is the first real
success that we have for good reads on emulsion-generated beads. The signal is really good for nearly all of
the cycles.
Color-coding here is the
same as described above:
T1 CACACACACACACACACTCCACCACT
T2 GTGTGTGTGTGTGTGTCCACCACTCT
T3 AGTGCTCACACACGTGATCCACCACT
T4 CAGCCGAACGACCGATCCACCACTCT
T5 ATGTGAGAGCTGTCGTCCACCACTCT
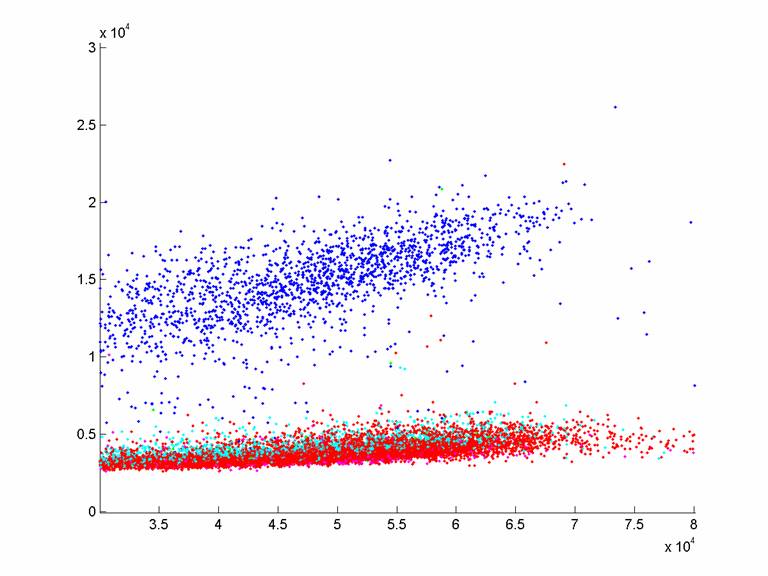
Sample graph from the same
primer as the sample graph above, 37C.8N.CG, sequencing bases 10,11,12 on T4
(except with emulsion-beads instead of loaded-beads, and showing only
well-amplified, clonal beads). Note the
signal on these beads is actually higher than the loaded beads, which is a
pleasant surprise. Some reasons why this
could be include (a) more template on amplified beads than has been the case in
the past with Greg’s optimizations, (b) switch to Sequenase
from Klenow.
Lots
of potential things to try:
(1)
switch to four-color sequencing
(2)
repeat emulsion T1.T5 experiment with additional
primers at 10N,12N,14N and adjusted template ratios
(3)
full set of primers on a more complex library
(4)
hybrid primers with constant region, universal or
fixed bases, wobble bases, and fixed termini.
(5)
single-base-matched termini with Sequenase.
(6)
longer anchors to bring up T5 signal?
(7)
stabilizing primer hybridized to distal constant
region.
(8)
other natural and mutant polymerases