Update on PCR preparation of knockouts. New! 1 July 2000.
pKO3 & pKOV (new! 9 May 1999) Maps and Sequences
See also: Link, A.J., Phillips, D. and Church, G.M. (1997) Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: Application to open reading frame characterization. J. Bacteriology 179: 6228-6237.
E.coli in-frame gene deletion projects
**NEW** E. coli knockout and amplification primers
Back to the Church Lab Home Page
Excerpts
from Andy Link's 1994 Harvard University Thesis, "Experimental Tools for
the Analysis of Genomes"
The vector pKO3 integrates into the chromosome by homologous recombination creating a tandem duplication at the nonpermissive temperature. When shifted to the permissive temperature, the pSC101 replication origin in the host chromosome is detrimental, and the vector is excised from the chromosome. To select for the loss of vector sequence from the cell, the B. subtilis gene sacB was incorporated into the vector, since expression of sacB in the presence of sucrose is detrimental to E. coli.
Because the entire protocol can be done by replica plating onto different selectable mediums at different temperatures, large numbers of genes can be simultaneously replaced with knockout alleles. Unlike other methods used for gene replacements in E. coli using ColE1 plasmids in a polA1 background (Gutterson and Koshland, 1983) or transformation of linear DNA into recBC sbcB or recD strains (Jasin and Schimmel, 1984; Winans et al., 1985; Shevell et al., 1988), this protocol is performed directly in wild-type strains. Moreover, since the system is plasmid based, gene replacements are easily performed in any genetic background that is recombination proficient.
Two approaches were developed for generating mutant alleles in vitro which could be inserted into the pKO3 vector for replacing open reading frames on the E. coli chromosome. Both methods were developed with the goal of eventually applying them to the large number of unidentified open reading frames. The first set of tools inserts an antibiotic marker into randomly cloned sequences to disrupt the open reading frame. From this first method, a second approach was derived which uses crossover PCR to generate precise deletions of the open reading frames. These two methods were applied to two E. coli reading frames identified in the protein content survey of E. coli.
Andrew
J. Link, Dereth R. Phillips, and George M. Church
Harvard Medical School
Department of Genetics
77 Avenue Louis Pasteur
Boston, MA 02115
To receive the plasmid:
Please contact Addgene ( http://www.addgene.org/pgvec1). pKOV is available to scientists at academic and non-profit institutions through Addgene, the non-profit plasmid repository.
The following methods were developed for working with the gene replacement vector pKO3. The plasmid and gene replacements protocols were derived from Hamilton et al. (1989) (J. Bacteriology 171: 4617-4622).
At the nonpermissive temperature, altered chromosomal sequences carried on the pSC101 plasmid integrates into the chromosome by homologous recombination to create an imperfect tandem duplication. When the cells are shifted to the permissive temperature, the cointegrant tends to undergoes a second recombination event regenerating the plasmid in the cell. Depending on the site of the second recombination, either the wild-type or the mutant allele is left behind in the chromosome. To select for the loss of vector sequence from the cell, the B. subtilis gene sacB was incorporated into the vector, since expression of sacB in the presence of sucrose is detrimental to E. coli.
Mailing of pKOV: for any information about mailing plasmids, please contact Addgene
Strain: The gene replacement experiments used the recombination proficient strains EMG2 (F' lambda+) or MC1061 (F- araD139 del(ara leu)7696 del(lacY74) galU galK hsdr hsdM+ strA).
Media and growth conditions: All strains were grown in LB medium (1% (w/v) bactotryptone, 0.5% (w/v) yeast extract, 0.5% (w/v) NaCl) with the appropriate selection. For antibiotic selection, the concentration of chromamphenicol was 20 mg/ml. For selection against sacB, LB medium was supplemented with sucrose to a final sucrose concentration of 5%(w/v)
NOTE: pKO3 has a temperature sensitive pSC101 replication origin. To recover the plasmid, strains harboring the plasmid must be grown at 30 deg C under chloramphenicol selection.
Isolation of pKO3: E. coli strains harboring pKO3 plasmid are grown in LB media at 30 deg C under chloramphenicol selection to stationary phase. Plasmid DNA is isolated usually the alkaline-lysis method (Birnboim and Doly) or Qiagen's plasmid prep kit.
For an analytical restriction digest of pKO3, 1.5 ml of overnight culture is used in the mini-prep, and the recovered plasmid DNA is resuspended in 25 ul of TE. 5 ul of the plasmid DNA is used for the restriction digestion and detection on ethidium bromide stained agarose gels. For preparing preparative amounts of pKO3, 50 to 1000 ml of overnight culture are used. The pSC101 plasmid is present at 5-10 copies per cell.
Electroporation: 40 ml of electroporation competent cells (1x10^11 cells/ml) were mixed with 1-3 ml of DNA in a ice-cold 500 ul microfuge tube and transferred to a 0.2 cm electroporation cuvette (Biorad, Inc). The cells are electroporated at 2,500 kV with 25 microfarad and 200 ohm resistance. Immediately after electroporation, 1 ml of SOC (2% bactotryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose) media is added to the cuvette. The cell are transferred to a 17 x 100 mm polypropylene tube and allowed to recover for 1 h at 30 deg C shaken at 250 rpm before plating on selective media.
DNA sequencing: Sequencing the left and right vector-insert junctions of inserts cloned into the BamHI site of pKO3 used the sequencing primers pK03-L and pK03-R. Cycle sequencing was performed essentially as described (Murray, 1989) using the Stratagene "Cyclist Sequencing" kit (Stratagene, Inc.) and plasmid DNA isolated using a Qiagen plasmid prep kit. Sequencing products were labeled with alpha-32P-dATP.
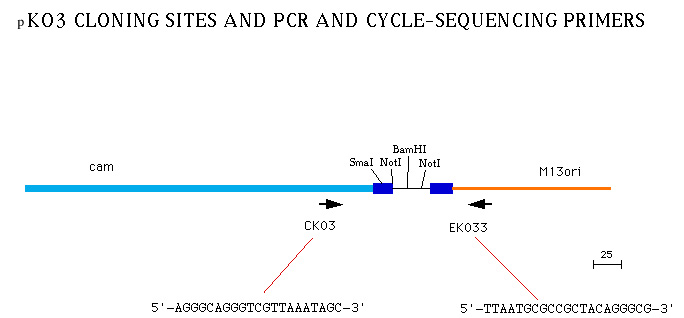
pK03-L: 5'-AGGGCAGGGTCGTTAAATAGC-3'
pK03-R: 5'-TTAATGCGCCGCTACAGGGCG-3'.
Polymerase chain reaction: Primers pK03-L and pK03-R were used for PCR amplifying inserts cloned into the BamHI site of pKO3. The PCR reactions used either purified DNA or bacterial colonies as the starting template. All PCR reactions were performed in a Perkin-Elmer 9600 thermal cycler. PCR reaction buffer (Ponce and Micol (1992), NAR 20: 623) consisted of 30 mM tricine (pH 8.4), 2 mM MgCl2, 5 mM beta-mercaptoethanol, 0.01% w/v gelatin, 0.1% w/v Thesit, 200 uM each dNTP, 600 nM of each primer, and 1 unit Taq polymerase (Boehringer Mannheim). The PCR reaction mixture was denatured at 94 deg C for 3 min before adding the Taq polymerase. The thermal cycle profile was 15 sec at 94 deg C, 15 sec at 55 deg C, and 30 sec at 72 deg C. All experiments used 30 cycles, and a final 5 min 72 deg C hold step.
Gene replacement: Mutant alleles cloned into the pKO3 gene
replacement vector are electroporated into
recombination proficient strains (eg. EMG2) and
allowed to recover for 1 h at 30 deg C. The cells are plated
on prewarmed chloramphenicol/LB
plates and incubated at 42 deg C. To measure the integration frequency, the electroporated cells are also plated
on chloramphenicol/LB plates at 30 deg C. From the 42
deg C plate, 1-5 colonies are picked into 1 ml of LB broth, serially diluted,
and immediately plated at 30°ree;C on either 5% w/v sucrose or 5% sucrose+antibiotic plates. The 5% sucrose plates are
replica plated to chloramphenicol plates at 30 deg C
to test for loss of the replacement vector (cms). The
gene replacement is confirmed by either PCR using primers flanking the targeted
open reading frame or by genomic Southern's.
IMPORTANT THINGS TO REMEMBER:
1) pK03 has a temperature sensitive Psc101 replication orgin. Strains harboring the plasmid must be grown at 30 deg C under chloramphenicol selection.
2) SacB appears to be mildly stressful to the cell even without added sucrose. It is a good idea not to carry the plasmid for too many generations in a single strain as you will accumulate mutations either in the sacB gene or in the bacterial genome to bypass this toxicity.
3) Make sure you test your plasmid for all of the markers (ts, sacB, cat) before you clone anything into it. We generally check each batch of pK03 before shipping. In the absence of selection the ts and sacB markers are basically stable. The pK03 strain has "very-slow-growth" (NOT "no-growth") at 43 deg or on sucrose, so streak tests are not recommended. Instead, we take a pK03 "test-colony" and serially dilute and plate for single colonies at both 30 and 40 degrees. If the 43 degree colonies are smaller than the 30 degree colonies then the "test-colony" is "ts". When performing the knock-out protocol, we only pick the large colonies on the 43 deg plates. We do the same serial dilution with sucrose/LB versus LB plates to test for SacB.
4) We are not prepared to advise researchers on the use of this plasmid in any other species or using any cloning sites other than those used in the J. Bact. Paper. All other uses are at you won risk.
5) The pSC101 plasmid is considered to be low copy and is present at 5-10 copies per cell. You may need to adjust your plasmid prep protocols to increase your yield.
For additional updates, vector and tag sequences and information about E. coli community gene knockout resource sharing please consult our web site: http://arep.med.harvard.edu/gmc/ecoko.html
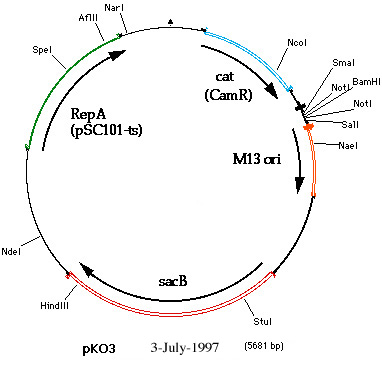
Figure
1 (pKO3_map_3-jul-97.jpg): A map of pKO3 showing the
unique restriction sites. The sequences \used to construct the map have not
been verified. With the exception of BamHI, NotI, and HindIII, the
restriction sites have not been confirmed.
Figure 2(pKO3_CLONING_SITE.jpg): A detailed map of the BamHI cloning site used for cloning genomic inserts for gene replacement experiments in the Church lab. pK03-L and pK03-R are primer sequences used for PCR amplification across the cloning site and cycle sequencing the vector-insert junctions.
Sequence files (GCG format): (pKO3 vector) (pKOV
{kind=link}
{kind=link}