HUMMUS
Introduction
The recent availability of various draft sequences of the assembled human & mouse genomes has opened the possibility of comprehensive searches for stretches of DNA exhibiting significant conservation over evolutionary time. We adopted a BLAST-based strategy using the Celera draft mouse genome assembly and the NCBI draft human genome assembly to define ~1.15 million “islands” of strong homology, covering the most conserved ~10% of each genome. We have used these elements to generate a “humanized” version of the mouse genome and a “mousenized” version of the human genome. We are working towards making all of these resources publicly available in the near future. In the meantime, we offer from this page: (1) statistics on the current build of HUMMUS; (2) graphical representations of mouse-human conservation in the context of exon-intron mappings of mRNA sequences to genomic sequence.
Example of mRNA ® genome mapping:


The x-axis represents genomic sequence. The green and blue boxes of the upper row represent individual exons of an mRNA mapped to genomic sequence with the SIM4 tool. Coding portions of the mRNA are colored blue and non-coding portions (e.g. the 5’ and 3’ UTRs) are colored green. In this case, the transcript proceeds from left-to-right along the contig. In the lower row (orange), the height of the vertical bar at each position is proportional to the level of mouse-human conservation over a 50-base-pair window of the overlaid human genome, centered at that position. That HUMMUS consists of a set of “islands” of strong conservation, rather than a continuous full alignment, can be clearly seen (see Methods). As expected, the positions of the coding exons correlate strongly with spikes in mouse-human conservation. Also notable are the stretches of conservation upstream and downstream of the transcript, as well as in its 3’ UTR.
Statistics
HUMMUS is available in three forms:
![]() islands of
conservation a set of ~1.15 million gapped alignments of
streches of syntenic conserved mouse & human sequence
islands of
conservation a set of ~1.15 million gapped alignments of
streches of syntenic conserved mouse & human sequence
![]() “mousenized” human
genome an overlay of a reference assembled human genome with corresponding
mouse sequence
“mousenized” human
genome an overlay of a reference assembled human genome with corresponding
mouse sequence
![]() “humanized” mouse
genome an overlay of a reference assembled mouse genome with
corresponding human sequence
“humanized” mouse
genome an overlay of a reference assembled mouse genome with
corresponding human sequence
We present here some basic statistics on the “islands of conservation” and the “mousenized” human genome.
![]() islands of
conservation
islands of
conservation
Number of alignments 1,150,000
Average alignment size 254.6 bp
Estimated fraction of false-positive bases ~0.0004
Estimated coverage ~90%
 “mousenized” human genome
“mousenized” human genome
Number of overlaid bases 288,384,392 bp (~10% of genome)
Overall % nucleotide identity for overlaid regions 75.3%
Overall % gapped bases in overlaid regions ~2%
To obtain an independent measure of coverage and to calculate statistics of conservation over known genes, we mapped 8,100 RefSeq transcripts
with defined ORFs back to the “mousenized” human genome with the MEGABLAST and SIM4 tools (see methods). 7,442 of these transcripts intersected with HUMMUS-defined overlaid bases (~92%; consistent with our independent estimate of ~90% coverage).
The following statistics were observed for the 7,442 covered mRNA sequences:
% of coding bases overlaid with mouse sequence 93.0%
% overall nucleotide identity of overlaid coding bases 84.2%
% of 5’ UTR bases overlaid with mouse sequence 48.2%
% overall nucleotide identity of overlaid 5’ UTR bases 78.6%
% of 5’ UTR bases overlaid with mouse sequence 49.3%
% overall nucleotide identity of overlaid 5’ UTR bases 77.3%
73% of the transcripts had 3’ UTRs with 100+ bp of aligned bases
Methods
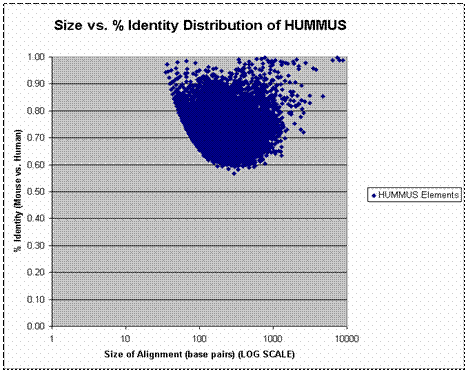
We chose to adopt BLAST as a sequence alignment algorithm for our initial attempts at genome vs. genome alignment because of its established statistical foundation. BLAST is a heuristic algorithm that speeds dynamic programming based alignment by requiring perfect match “seed” (e.g. the word size parameter) to initiate alignments. Reducing the word-size increases the sensitivity of BLAST to lower % identity alignments but increases the computational time required to execute the alignment. It is computationally intractable to blast the full human genome against the full mouse genome with sensitive parameters. We therefore adopted a two-step strategy that took advantage of the extensive synteny between the human and mouse genomes. In the first step, syntenies were defined by applying the MEGABLAST tool to align ~18 million Celera raw mouse genome shotgun reads to a reference human genome with relatively insensitive parameters (word size of 16). An assembled version of the Celera mouse genome became available in August 2000, and we proceeded to map these alignments back to the assembled mouse genome. Our ‘low-resolution’ alignments, though far from comprehensive, constituted a set of defined syntenies that were exploited to reduce the total amount of blasting that we had to perform. We proceeded to focus on 50 kb chunks of sequence centered on these positions of predicted synteny. A version of the human genome that was pre-masked for repetitive elements and low-complexity sequence was used to avoid spurious alignments. Putatively syntenic mouse and human chunks were blasted against one another using highly sensitive parameters (word size of 7). A full set of non-overlapping alignments was generated in this manner. By blasting chunks of non-syntenic mouse and human sequence against one another with the same parameters, we were able to estimate a false positive rate (e.g. the fraction of bases that are aligned for reasons other than synteny) of ~0.0004. The graph below is presented to give a sense of the % identity and lengths of alignments that are captured by the BLAST parameters that we used. Each point on the graph corresponds to a single HUMMUS alignment (e.g. and “island” of conservation). The x-axis (log-scale) indicates the size of individual alignments. The y-axis indicates the % identity of corresponding alignments.
 |
The NCBI UniGene resource is the result of the partitioning of millions of ESTs and tens-of-thousands of mRNA into a “non-redundant, gene-oriented” set of clusters. As part of the resource, a single sequence is selected from each cluster as its longest, highest-quality member. We refer to these sequences as the Best-Of-UniGene (BOU) sequences. Many of the BOUs are full-length mRNAs (often the same mRNAs found in the RefSeq resource, though this is not always the case). We used the MEGABLAST tool to map approximate genomic locations and the SIM4 tool to obtain more precise mappings of each human and mouse UniGene BOU sequence onto genomic coordinates of the overlaid human and mouse genomes, respectively. Graphical representations integrating information on exon-intron structure and mouse-human conservation were generated for each UniGene cluster. The map above (see Introduction section) is an example. A full set of graphical representations for all UniGene clusters that we were able to map is available:
Related Links
Computational Discovery of Overlapping Transcriptional Units in the Human and Mouse Genomes
Related References
- Shendure, J. and Church, GM. Computational Discovery Of Bidirectionally Transcribed Regions of the Human & Mouse Genomes. (submitted)
- Florea L., Hartzell G., Zhang Z., Rubin GM., Miller W. Genome Res 8, 967-74 (1998).
- Zhang Z., Schwartz, S., Wagner, L., Miller, W.A. J Comput Biol 7, 203-14 (2000).
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. J Mol Biol 215, 403-10 (1990).
- Lander E.S. Nature. 2001 Feb 15;409(6822):860-921.
- Venter J.C. et al. Science. 2001 Feb 16;291(5507):1304-51.
Contact Information
Jay Shendure
jay_shendure@student.hms.harvard.edu
Last revised : November 11, 2003